- Гідрологія і Гідрометрія
- Господарське право
- Економіка будівництва
- Економіка природокористування
- Економічна теорія
- Земельне право
- Історія України
- Кримінально виконавче право
- Медична радіологія
- Методи аналізу
- Міжнародне приватне право
- Міжнародний маркетинг
- Основи екології
- Предмет Політологія
- Соціальне страхування
- Технічні засоби організації дорожнього руху
- Товарознавство продовольчих товарів
Тлумачний словник
Авто
Автоматизація
Архітектура
Астрономія
Аудит
Біологія
Будівництво
Бухгалтерія
Винахідництво
Виробництво
Військова справа
Генетика
Географія
Геологія
Господарство
Держава
Дім
Екологія
Економетрика
Економіка
Електроніка
Журналістика та ЗМІ
Зв'язок
Іноземні мови
Інформатика
Історія
Комп'ютери
Креслення
Кулінарія
Культура
Лексикологія
Література
Логіка
Маркетинг
Математика
Машинобудування
Медицина
Менеджмент
Метали і Зварювання
Механіка
Мистецтво
Музика
Населення
Освіта
Охорона безпеки життя
Охорона Праці
Педагогіка
Політика
Право
Програмування
Промисловість
Психологія
Радіо
Регилия
Соціологія
Спорт
Стандартизація
Технології
Торгівля
Туризм
Фізика
Фізіологія
Філософія
Фінанси
Хімія
Юриспунденкция
Кінетика ферментативних реакцій
Ферментативна кінетика досліджує вплив на швидкість перебігу реакції різних хімічних речовин і фізико-хімічних чинників, які є достатньо численними та різноманітними. До них відносять концентрацію фермента та субстрату, рН і температуру, наявність активаторів або інгібіторів. Вивчення кінетики ферментативної реакції важливо для вибору одиниць активності ферментів, здійснення їх очищення, планування проведення досліджень та інтерпретації результатів тощо.
1.4.1. Вплив концентрації фермента та субстрату на швидкість ферментативної реакції.Швидкість будь-якої ферментативної реакції залежить від концентрації фермента. Упереважній більшості випадків у початковий період реакції, за умови надлишку фермента та невеликої кількості продукту швидкість реакції (V) прямо пропорційна його концентрації – [Е] і має лінійний характер: V=К×[Е], де К – коефіцієнт (рис. 5.4, а). Але з часом кількість продукту зростає і з’являється можливість для перебігу зворотної реакції, внаслідок чого лінійна залежність втрачається.
Якщо ж концентрацію фермента залишити постійною, змінюючи лише концентрацію субстрату [S], то графік швидкості ферментативної реакції буде описуватися гіперболою (рис. 1.6, Б).

Рис. 1.6. Залежність швидкості ферментативної реакції від концентрації фермента (А) та субстрату (Б)
При збільшенні кількості субстрату початкова швидкість зростає і коли фермент повністю насичується субстратом, тобто відбувається максимально можливе утворення фермент-субстратних комплексів, спостерігають найвищу швидкість утворення продукту. Але подальше збільшення концентрації субстрату не призведе до збільшення утворення продукту, оскільки швидкість реакції збільшуватися не буде. Описаний стан відповідає максимальній швидкості реакції (V max).
На основі аналізу залежності V від [S] Л. Міхаеліс і М. Ментен сформулювали в 1913 р. загальну теорію кінетики дії ферментів. Вони постулювали, зокрема, що ферментативна реакція є двостадійною. На першій стадії фермент вступає в швидку зворотну взаємодію з субстратом з утворенням фермент-субстратного комплексу (ES), а під час другої стадії, яка відбувається повільніше і лімітує швидкість процесу, комплекс ES розпадається з утворенням продукту реакції (Р) та відновленого стану ферменту:

де к1 – константа швидкості утворення ES, к-1 – константа швидкості зворотної реакції (розпаду ES), к2 – константа швидкості утворення продукту реакції. Співвідношення констант швидкостей (к-1 + к2) / к1 називають константою Міхаеліса (КМ).
На основі цього було виведено рівняння, яке пов'язує V і [S], відоме під назвою рівняння Міхаеліса:
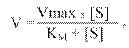
де: V – початкова швидкість реакції, тобто швидкість, що реєструється впродовж періоду часу, за який рівень субстрату не перевищує 10 %. У цей період швидкість реакції можна вважати приблизно постійною, оскільки, по-перше, зменшення кількості субстрату невелике, а по-друге, концентрація продукту незначна. Vмакс дає характеристику каталітичній активності фермента і має розмірність швидкості ферментативної реакції моль/л, тобто, вона визначає максимальну можливість утворення продукту при певній концентрації фермента в умовах надлишку субстрату.
У випадку, коли швидкість реакції рівна половині максимальної (V=Vmax/2), то КМ = [S]. Таким чином, константа Міхаеліса чисельно дорівнює концентрації субстрату, при якій швидкість ферментативної реакції становить половину від максимальної. Ця величина характеризує спорідненість того чи іншого фермента до конкретного субстрата і є величиною постійною, незалежною від концентрації фермента. Якщо КМ значно більша від [S], тобто Км >> S, то сума (КМ + S) приблизно дорівнює КМ, відповідно рівняння набуває вигляду: V = Vмакс × [S] / КМ. у цьому випадку швидкість ферментативної реакції прямо пропорційна концентрації субстрату, тобто при малих концентраціях субстрату швидкість буде зростати із збільшенням концентрації. Якщо [S] >> КМ, то зростання концентрації субстрату на величину КМ + [S] практично не впливає і нею можна знехтувати. Тому швидкість реакції буде дорівнювати максимальній швидкості: V=Vмакс.
 Обробка рівняння Міхаеліса-Ментен за методом подвійних зворотних величин дає змогу відобразити залежність V від [S] прямою лінією (рівняння Лайнуївера-Берка) (рис. 5.5).
Обробка рівняння Міхаеліса-Ментен за методом подвійних зворотних величин дає змогу відобразити залежність V від [S] прямою лінією (рівняння Лайнуївера-Берка) (рис. 5.5).

Між 1/V та 1/[S] є прямопропорційна залежність, яка дозволяє легко отримати значення кінетичних констант КМ та Vмакс, що неможливо при аналізі звичайної гіперболи.
Із рівняння та графіка випливає, що кутовий коефіцієнт прямої (tg кута нахилу) дорівнює Км/Vмакс. Значення цих констант легко знайти за графіком.
1.4.2. Залежність швидкості ферментативної реакції від температури.Зростання температури до певних визначених меж чинить вплив на швидкість ферментативної реакції, подібно до впливу температури на будь-яку хімічну реакцію, що супроводжується прискоренням руху молекул і, відповідно, прискоренням ймовірності взаємодії реагуючих речовин. Крім того, температура може підвищувати енергію реагуючих речовин, що теж прискорює реакцію. Однак, швидкість ферментативної реакції має свій температурний оптимум, перевищення якого супроводжується зниженням ферментативної активності внаслідок термічної денатурації білкових молекул (рис. 1.8).
Для більшості ферментів людини оптимальна температура 37 – 40 °С, проте в природі існують і термостабільні ферменти: Тaq-полімераза, виділена з мікроорганізмів, яка не інактивується навіть при 95 °С. Цей фермент використовують у науково-практичній медицині для молекулярної діагностики захворювань із використанням методу ланцюгової полімеразної реакції.
Зниження температури нижче оптимальної тимчасово сповільнює активність ферменту внаслідок зменшення процесів дифузії молекул; повернення того чи іншого фермента в оптимальне температурне середовище відновлює його активність. Цю здатність ферментів широко використовують у медицині для пригнічення метаболічних процесів у тканинах (під час трансплантації органів, операцій на серці), у фармації для збереження препаратів і лікарських форм (наприклад, білкових препаратів, відварів, настоїв, емульсій тощо), у народному господарстві, наприклад, для збереження харчових продуктів.
1.4.3. Залежність швидкості ферментативної реакції від pH середовища. Для кожного фермента існує певне значення рН середовища, в якому він виявляє максимальну активність. Для пепсину воно становить 1,5 – 2,0, піруваткарбоксилази 4,8, аргінази – 9,5 – 10,0. Проте, більшість ферментів організму людини мають оптимум рН, наближений до нейтрального: фумараза 6,5; каталаза 6,8 – 7; уреаза 6,8 – 7,2; амілаза слини 6,8 – 7,4, карбоксипептидаза 7,5; трипсин 7,5 – 8,5, (рис. 1.9).
 Вплив рН на активність фермента пов'язаний із іонізацією функціональних груп амінокислотних залишків білкової молекули, що забезпечує оптимальну конформацію активного центра. Відхилення рН від оптимальних величин порушує іонізацію функціональних груп в активному центрі фермента. Так, наприклад, залуження середовища спричинює від’єднання іонів Н+ від карбоксильних груп (СОО-), тоді як закислення, навпаки, приєднання протонів до вільних аміногруп (NН3+). Це викликає зниження активності ферменту, порушує спорідненість субстрату до фермента і гальмує каталітичний процес в цілому. При значному відхиленні від оптимального значення рН може відбуватися денатурація білкової молекули з повною втратою ферментативної активності. Зміна рН середовища може також впливати і на просторову організацію субстрату.
Вплив рН на активність фермента пов'язаний із іонізацією функціональних груп амінокислотних залишків білкової молекули, що забезпечує оптимальну конформацію активного центра. Відхилення рН від оптимальних величин порушує іонізацію функціональних груп в активному центрі фермента. Так, наприклад, залуження середовища спричинює від’єднання іонів Н+ від карбоксильних груп (СОО-), тоді як закислення, навпаки, приєднання протонів до вільних аміногруп (NН3+). Це викликає зниження активності ферменту, порушує спорідненість субстрату до фермента і гальмує каталітичний процес в цілому. При значному відхиленні від оптимального значення рН може відбуватися денатурація білкової молекули з повною втратою ферментативної активності. Зміна рН середовища може також впливати і на просторову організацію субстрату.
1.4.4. Регуляція ферментативних процесів.Зазвичай кожен метаболічний шлях має свої ключові ферменти, які називають регуляторними, оскільки завдяки їм відбувається регуляція швидкості всього шляху. Ці ферменти можуть каталізувати початкові, або незворотні або найповільніші реакції, вони також розташовуються в точках розгалуження метаболічного шляху. Впливаючи на такі ферменти модифікаторами (активаторами чи інгібіторами) можна змінити швидкість перебігу не лише однієї реакції, а й усього метаболічного шляху.
Будь-які зміни оточуючого чи внутрішнього середовища вимагають включення адаптаційних процесів, що реалізуються, першою чергою, через зміну швидкості тої чи іншої ферментативної реакції.
Регуляція швидкості ферментативної реакції здійснюється трьомах шляхами: зміною кількості ферменту; доступністю субстрату та кофермента; зміною каталітичної активності молекули фермента.
Перший шлях регуляції є механізмом довготривалої адаптації ферментів.Для його включення і повної реалізації необхідно декілька годин або діб. Він полягає у зміні впливу на систему ядерного геному або рибосомального білкового синтезу (вплив на процеси транскрипції та трансляції)метаболітів, гормонів та інших біологічно активних речовин. Цей тип регуляції характерний здебільшого для мікроорганізмів, ферменти яких поділяють на два класи: конститутивні, які синтезуються мікроорганізмами постійно, не залежно від умов існування та адаптивні, інтенсивність біосинтезу яких змінюється залежно від змін умов існування. Адаптивні ферменти мікроорганізмів поділяють на індуцибельні та репресибельні, тобто такі, активність синтезу яких підвищується або гальмується залежно від дії певних сполук-ефекторів.
Другий шлях регуляції здійснюється на рівні двох параметрів: субстрату та кофермента. Чим більша концентрація субстрату (особливо першого, вихідного), тим вища швидкість метаболічного шляху. Стосовно кофермента слід зазначити, що важливе значення для регуляції має наявність регенерованих коферментів. Наприклад, у реакціях дегідрування коферментами дегідрогеназ слугують окиснені форми НАД+, ФАД, ФМН, які відновлюються в ході реакції. Для того, щоб коферменти могли знову брати участь у реакції, необхідна їх регенерація, тобто перехід в окиснену форму.
Третій шлях регуляції активності ферментів може відбуватися за чотирма основними механізмами (L. Stryеr, 1995): ковалентною модифікацією; обмеженим протеолізом; білок-білковими взаємодіями; алостерично.
До регулюючих механізмів можна віднести і явище компартменталізації – строга локалізація ферментів у різних органелах, що дозволяє одночасно перебігати різноспрямованим процесам (наприклад, синтез і розпад) у межах однієї клітини. Так, процес синтезу жирних кислот відбувається в цитоплазмі, а їх розпад зосереджений у мітохондріях.
Ковалентна модифікація ферментів – один із механізмів контролю метаболічних процесів, що може відбуватися шляхом зворотного фосфорилування-дефосфорилування, метилування, аденілування, АДФ-рибозилування білків-ферментів. Фосфорилують білки спеціальні ферменти протеїнкінази, які за допомогою залишку фосфату АТФ здійснюють фосфорилування серинового, тирозинового чи треонінового радикалу відповідного білка. Зворотну реакцію – дефосфорилування білків – каталізують протеїнфосфатазами. Субстратами протеїнкіназ є численні ферментативні білки (глікогенфосфорилаза, кіназа фосфорилази b, глікогенсинтаза, тригліцеридліпаза, піруватдегідрогеназа, ацетил–КоА-карбоксилаза тощо), деякі білки мембранних каналів, гістони хроматину тощо. Приєднання залишка фосфорної кислоти призводить до зміни конформації активного центра, при цьому результат може бути двояким: фосфорилування багатьох білків-ферментів трансформує їх у каталітично активну форму (фосфорилування глікогенфосфорилази, кінази фосфорилази b, тригліцеридліпази тощо), тоді як фосфорилування інших ферментативних білків (глікогенсинтази, β–ГОМК-редуктази) є, навпаки, механізмом їх інактивації (рис. 1.10).

Рис. 1.10. Регуляція активності ліпази
Зміна активності фермента внаслідок фосфорилування зазвичай зворотна і регулюється гормонами, що дозволяє швидко змінювати активність ключових ферментів метаболічних шляхів.
Активація ферментів шляхом обмеженого протеолізу. Активація низки ферментів (здебільшого травного тракту або плазми крові) може відбуватися протеолітичним шляхом. Такі ферменти синтезуються в неактивній формі у вигляді проферментів або зимогенів (попередників ферментів), активний центр яких замаскований додатковою ділянкою пептидного ланцюга, внаслідок чого субстрат не може взаємодіяти з активним центром. Наприклад, проферментом пепсину є пепсиноген, який синтезується головними клітинами шлункових залоз. Відщеплення від його молекули невеликого пептидного ланцюга за участі хлоридної кислоти шлункового соку призводить до утворення пепсину та формування його активного центра. Профермент трипсиноген, який утворюється в підшлунковій залозі, у дванадцятипалій кишці під впливом ферменту ентерокінази шляхом відщеплення гексапептиду перетворюється на трипсин. Після цього створюються умови, які сприяють утворенню активного центру ферменту, і трипсиноген перетворюється на трипсин. Ці процеси можуть також відбуватися аутокаталітично, тобто під впливом вже утвореного пепсину чи трипсину відповідно.
Активація шляхом білок-білкових взаємодій включає в себе два механізми: активацію в результаті приєднання регуляторних білків і зміну каталітичної активності внаслідок асоціації чи дисоціації протомерів фермента.
До регуляторних білків, які можуть чинити вплив на активність ферментів належать:
- кальмодулін-кальційвмісний білок, який, зв'язуючись з чотирма іонами кальцію, сприяє збільшенню цитозольної концентрації останнього при певних біохімічних та фізіологічних процесах у клітині. Утворений комплекс кальмодулін-кальцій здатний до активації багатьох ферментів, зокрема фосфодіестерази циклічних нуклеотидів, кінази легких ланцюгів міозину тощо;
- протеїназні інгібітори блокують активність тканинних протеїназ – ферментів, здатних розщеплювати власні білки організму. Найактивнішими протеїназами є α2 – макроглобулін та α1 – антитрипсин (α1- протеїназний інгібітор), які блокують активність серинових та інших протеїназ;
- антигемофільний глобулін А (фактор VІІІ згортальної системи крові). Цей білок бере участь в активації фактора Х, який запускає весь коагуляційний каскад, що призводить до утворення тромба.
 Прикладом регуляції каталітичної активності шляхом асоціації/дисоціації протомерів може слугувати регуляція активності протеїнкінази А, функцією якої є фосфорилування інших білків-субстратів, що призводить до значного посилення регуляторного сигналу (каскадна система регуляції). Цей фермент в неактивній формі є тетрамером R2С2 який складається з двох регуляторних (R) і двох каталітичних (С) субодиниць (рис. 1.11).
Прикладом регуляції каталітичної активності шляхом асоціації/дисоціації протомерів може слугувати регуляція активності протеїнкінази А, функцією якої є фосфорилування інших білків-субстратів, що призводить до значного посилення регуляторного сигналу (каскадна система регуляції). Цей фермент в неактивній формі є тетрамером R2С2 який складається з двох регуляторних (R) і двох каталітичних (С) субодиниць (рис. 1.11).
Регуляторні субодиниці мають центри зв’язування для цАМФ (по 2 на кожну субодиницю). Активна протеїнкіназа представлена субодиницями С, для вивільнення яких необхідна дисоціація комплексу. Активація ферменту відбувається за участі цАМФ, який приєднуються до субодиниць R і змінює конформацію білка, що призводить до порушення комплементарності субодиниць R і С і дисоціації комплексу.
R2С2 + 4 цАМФ → 2 С + 2 (R-цАМФ)
Такий механізм регуляції зворотний. Від’єднання молекул цАМФ від регуляторних субодиниць призведе до асоціації регуляторних і каталітичних субодиниць протеїнкінази А з утворенням неактивного комплексу.
Алостерична регуляція ферментів. Активування ферментів може здійснюватися шляхом приєднання до алостеричного центра фермента специфічної модифікуючої групи (активатора), що сприяє зміні конформації фермента, його активного центра та, як наслідок, прискоренню ферментативної реакції(алостерична активація). Вона, основним чином, характерна для олігомерних ферментів, які складаються з кількох протомерів або мають доменну структуру. У центральних метаболічних шляхах вихідні речовини можуть слугувати активаторами ключових ферментів, при цьому алостеричній активації підлягають ті ферменти, які каталізують ключові реакції заключних етапів метаболічного шляху:

У якості прикладу розглянемо гліколіз – специфічний шлях розпаду глюкози: при утворенні надмірної кількості фруктозо-1,6-дифосфату спостерігається алостерична активація фермента піруваткінази (рис. 1.12).
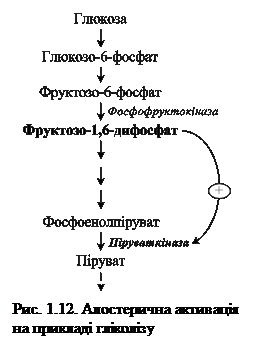 Роль активаторів можуть виконувати також органічні речовини (жовчні кислоти посилюють дію ліпази підшлункової залози) та неорганічні (іони хлору є активаторами амілази слини; іони водню посилюють активність пепсину). Проте існують випадки, коли одна й таж речовина виступає активатором для одного фермента, а інгібітором – для іншого.
Роль активаторів можуть виконувати також органічні речовини (жовчні кислоти посилюють дію ліпази підшлункової залози) та неорганічні (іони хлору є активаторами амілази слини; іони водню посилюють активність пепсину). Проте існують випадки, коли одна й таж речовина виступає активатором для одного фермента, а інгібітором – для іншого.
Іони металів часто слугують специфічними активаторами для низки ферментів, оскільки вони можуть бути компонентами активного центра, сприяють взаємодії субстрату з ферментом, беруть участь у формуванні третинної структури фермента (залізо в складі фермента каталази, мідь у складі аскорбатоксидази). Це стосується, в основному, катіонів. Аніони можуть позитивно впливати на ферментативну реакцію, прискорюючи її другий етап.
Інгібування ферментів.Дія багатьох ферментів може бути загальмована, а в низці випадків і повністю припинена під впливом певних хімічних речовин – інгібіторів. Останні за тією або іншою причиною частково або повністю перешкоджають утворенню активного фермент-субстратного комплексу. Зокрема, токсичність багатьох отрут для живих організмів, лікувальні ефекти деяких лікарських речовин зумовлені їх інгібуючою дією на ферменти. Серед інгібіторів є як синтетичні речовини, так і природні метаболіти.
Процес інгібування ферментів може бути зворотнім і незворотнім. Якщо молекула інгібітора викликає стійкі зміни, модифікацію функціональних груп ферменту або їх руйнування, то такий тип інгібування називається незворотнім. Незворотне гальмування виникає, коли утворений комплекс фермент-інгібітор практично не дисоціює. Такі інгібітори хімічно модифікують важливі функціональні групи фермента, тому після усунення інгібітора шляхом діалізу активність модифікованого фермента не відновлюється. Незворотні інгібітори мають властивості клітинних отрут.
Прикладами незворотних інгібіторів є диізопропілфторфосфат (ДІФФ), який інактивує низку гідролаз (трипсин, хімотрипсин, ацетилхолінестеразу тощо), модифікуючи важливий для активності цих ферментів залишок серину; фторид натрію, який інгібує фосфатази, фенапролін – металовмісні ферменти.
| |
Зворотні інгібітори взаємодіють з ферментом без утворення ковалентних зв'язків. Після інкубації з утворенням комплексу фермент-інгібітор активність фермента відновлюється при видаленні інгібітора шляхом діалізу. Прикладами таких інгібіторів є клітинні метаболіти та їх структурні аналоги, які знижують активність ферментів.
За механізмом дії зворотні інгібітори ферментів поділяють на конкурентні, неконкурентні, безконкурентні, субстратні (або метаболічні) та алостеричні.
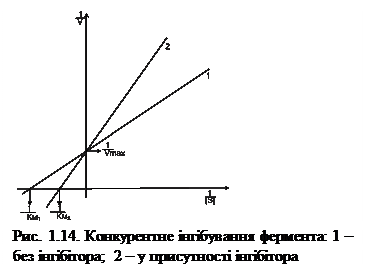
Конкурентне інгібування. Конкурентні інгібітори за будовою подібні до субстрату, вони конкурують із ним за зв’язування з активним центром фермента (рис. 1.13).
Характерною особливістю конкурентного гальмування є те, що ефективність інгібітора залежить від співвідношення концентрацій субстрату й інгібітора. При наявності субстрату [S] та інгібітора [І] одночасно відбуваться дві реакції:
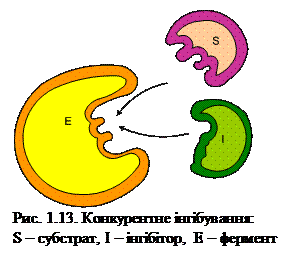

Гальмування відбудеться тоді, коли концентрація інгібітора перевищить концентрацію субстрату. У цьому випадку інгібітор утворює з ферментом комплекс фермент-інгібітор і виключає фермент із реакції. Але якщо концентрація субстрату буде вищою за концентрацію інгібітора, утворюється фермент-субстратний комплекс і дія інгібітора припиняється. Кінетичний аналіз за лайнуівером-Берком демонструє, що конкурентні інгібітори збільшують константу Міхаеліса Км ферменту і не впливають на максимальну швидкість реакції (рис. 1.14).
Прикладом конкурентного гальмування є дія малонової та деяких інших дикарбонових кислот на фермент сукцинатдегідрогеназу (СДГ), який каталізує в організмі перетворення янтарної кислоти (сукцинату) на фумарову (фумарат).

Малонова кислота є інгібітором даної реакції; у структурному відношенні вона подібна до янтарної кислоти і може конкурувати з останньою за місце в активному центрі СДГ (рис. 1.15).
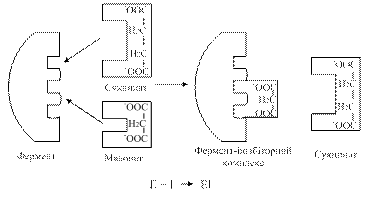
Рис. 1.15. Схема конкурентного гальмування СДГ малонатом
У цьому випадку СДГ ніби «ошукана»: вона замість субстрату (янтарної кислоти) захоплює його «двійника» (малонову кислоту) і субстрат уже не може розташуватися на контактній ділянці ферменту. Якщо ж навколо ферменту з'явиться багато молекул субстрату (янтарної кислоти), то субстрат витисне інгібітор з активного центру. Оскільки в комплексі фермент-інгібітор через специфічність дії ферменту хімічної реакції не відбувається, активність СДГ виявляється тільки відносно свого субстрату. Навпаки, якщо кількість молекул інгібітора є значною, то субстрату важче проникнути на контактну ділянку ферменту, активність його буде дуже низькою – ферментативна реакція блокується.
У якості інгібіторів ферментів за конкурентним механізмом у медичній практиці використовують речовини, які називають антиметаболітами. Будучи структурними аналогами природних субстратів, ці сполуки, з одного боку, викликають конкурентне інгібування ферментів, а з іншого – можуть використовуватися цими ж ферментами в якості псевдосубстратів, що призводить до синтезу аномальних продуктів, які не володіють функціональною активністю.
 У якості лікарських препаратів-антиметаболітів використовують аналоги нуклеотидів для лікування онкологічних захворювань, сульфаніламідні препарати (аналоги параамінобензойної кислоти (ПАБК)) для лікування інфекційних захворювань. ПАБК структурно подібна до сульфанілової кислоти, похідні якої є сульфаніламідними препаратами.
У якості лікарських препаратів-антиметаболітів використовують аналоги нуклеотидів для лікування онкологічних захворювань, сульфаніламідні препарати (аналоги параамінобензойної кислоти (ПАБК)) для лікування інфекційних захворювань. ПАБК структурно подібна до сульфанілової кислоти, похідні якої є сульфаніламідними препаратами.
 У мікроорганізмах в присутності ПАБК синтезується фолієва кислота, яка є важливим коферментом низки ферментів, що беруть участь у синтезі нуклеїнових кислот, а отже, і білків, тобто фолієва кислота є фактором росту бактерій, зокрема, стафілококів, пневмококів тощо. Цим забезпечується ріст і розмноження мікроорганізмів. У фолієвій кислоті ПАБК має два замісники: гетероциклічне похідне (R1) і глутамінову кислоту (R2). Сульфаніламідні препарати конкурують з ПАБК (структурна подібність) на стадії утворення фолієвої кислоти. Наявність сульфамідної групи в сульфаніламідних препаратах перешкоджає взаємодії ПАБК з глутаміновою кислотою, цим самим припиняється (блокується) біосинтез фолієвої кислоти. Гальмування синтезу фолієвої кислоти в мікроорганізмах призводить до порушення біосинтезу нуклеїнових кислот і білків, внаслідок цього пригнічується ріст і розмноження бактерій.
У мікроорганізмах в присутності ПАБК синтезується фолієва кислота, яка є важливим коферментом низки ферментів, що беруть участь у синтезі нуклеїнових кислот, а отже, і білків, тобто фолієва кислота є фактором росту бактерій, зокрема, стафілококів, пневмококів тощо. Цим забезпечується ріст і розмноження мікроорганізмів. У фолієвій кислоті ПАБК має два замісники: гетероциклічне похідне (R1) і глутамінову кислоту (R2). Сульфаніламідні препарати конкурують з ПАБК (структурна подібність) на стадії утворення фолієвої кислоти. Наявність сульфамідної групи в сульфаніламідних препаратах перешкоджає взаємодії ПАБК з глутаміновою кислотою, цим самим припиняється (блокується) біосинтез фолієвої кислоти. Гальмування синтезу фолієвої кислоти в мікроорганізмах призводить до порушення біосинтезу нуклеїнових кислот і білків, внаслідок цього пригнічується ріст і розмноження бактерій.
Таким чином, сульфанілова кислота та її N-заміщені аміди, виступають антиметаболітами ПАБК, зумовлюють бактеріостатичний ефект.
 Неконкурентним інгібуваннямназивають таке гальмування, при якому інгібітор не є структурним аналогом субстрату, він взаємодіє з ферментом або в ділянці, відмінній від активного центра, або з уже сформованим фермент-субстратним комплексом. Приєднання неконкурентного інгібітора викликає зміну конформацію молекули фермента в такий спосіб, що порушується взаємодія субстрату з активним центром фермента, що призводить до зниження швидкості реакції (рис. 1.16).
Неконкурентним інгібуваннямназивають таке гальмування, при якому інгібітор не є структурним аналогом субстрату, він взаємодіє з ферментом або в ділянці, відмінній від активного центра, або з уже сформованим фермент-субстратним комплексом. Приєднання неконкурентного інгібітора викликає зміну конформацію молекули фермента в такий спосіб, що порушується взаємодія субстрату з активним центром фермента, що призводить до зниження швидкості реакції (рис. 1.16).
У випадку утворення потрійного комплексу (фермент-субстрат-інгібітор) останній не спроможний перетворитися на продукт, у результаті чого реакція зупиняється.

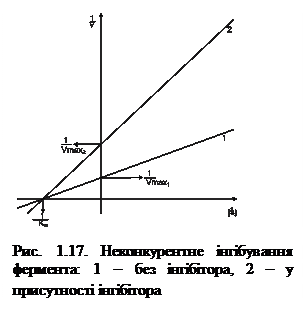 Неконкурентні інгібітори можуть бути проміжними продуктами метаболізму, які утворюються в живих організмах. Вони здатні зменшувати швидкість реакції (Vмакс), але не впливати на спорідненість ферменту до субстрату (Км) (рис. 1.17).
Неконкурентні інгібітори можуть бути проміжними продуктами метаболізму, які утворюються в живих організмах. Вони здатні зменшувати швидкість реакції (Vмакс), але не впливати на спорідненість ферменту до субстрату (Км) (рис. 1.17).
Неконкурентними інгібіторами є ціаніди, які міцно сполучаються з тривалентним залізом, яке входить в каталітичну ділянку гемінового фермента цитохромоксидази. Блокування останнього призводить до припинення тканинного дихання та загибелі клітини.
Велика токсичність іонів ртуті, свинцю, миш'яку обумовлена їх властивістю блокувати SH-групи каталітичних ділянок низки ферментів. Проте, важкі метали лише в невеликих концентраціях виконують роль неконкурентних інгібіторів, у великих кількостях вони є інактиваторами і діють як денатуруючі агенти. Для подолання інтоксикації, викликаної цими препаратами, застосовують різні реактиватори (або протиотрути), які витісняють інгібітор з комплексу. До них відносять, наприклад, SH-вмісні комплекси (цистеїн, унітіол тощо), лимонну кислоту, етилендіамінтетраацетатну кислоту тощо.
Безконкурентне інгібування.Цей тип інгібуванняспостерігають у тому випадку, коли інгібітор зворотно взаємодіє з ферментом тільки після утворення фермент-субстратного комплексу, тобто безконкурентний інгібітор не сполучається з ферментом у відсутності субстрату. Інгібітор полегшує приєднання субстрату, а потім, зв'язуючись, інгібує фермент. Це рідкісний вид інгібування.
Субстратне інгібування. Наявність надмірно високої концентрації субстрату викликає гальмування ферментативної реакції. Пояснюється цей факт тим, що молекули субстрату в надлишку займають неправильне положення в активному центрі фермента (рис. 1.18).

Рис. 1.18. Схема субстратного інгібування: А – молекула субстрату (S) оптимально розташована в активному центрі фермента (Е); Б – дві молекули субстрату зв’язалися неправильно з активним центром фермента
У багатьох біосинтетичних реакціях основним типом регуляції швидкості багатоступінчастого ферментативного процесу є алостеричне інгібування за типом зворотного зв'язку, коли кінцевий продукт, який за структурою подібний до субстрату, гальмує дію фермента. Такий тип інгібування доведений для всіх живих організмів і розглядається як один з основних типів регуляції активності ферментів і клітинного метаболізму взагалі, оскільки нагромадження надлишку продукта гальмує в подальшому його утворення.
Інколи алостерична регуляція виявляється у вигляді інгібування першого фермента біохімічних перетворень кінцевим продуктом:

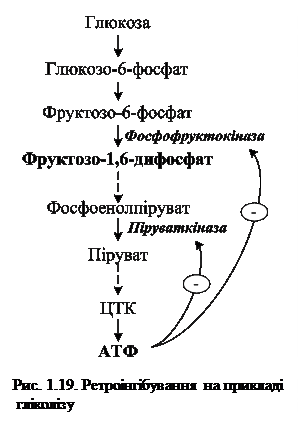 Фермент, який каталізує перетворення субстрату А на продукт Б має алостеричний центр для негативного ефектора (інгібітора), який виступає кінцевий продукт метаболічного шляху (Д). Якщо концентрація останнього збільшується, то інгібується активність одного з початкових ферментів (Е1). Така регуляція за принципом зворотного зв'язку (ретроінгібування) дозволяє контролювати вихід кінцевого продукту. Наприклад, при надлишку в клітині АТФ, яка утворюється під час гліколізу, відбувається ретроінгібування алостеричних ферментів фосфофруктокінази та піруваткінази (рис. 1.19).
Фермент, який каталізує перетворення субстрату А на продукт Б має алостеричний центр для негативного ефектора (інгібітора), який виступає кінцевий продукт метаболічного шляху (Д). Якщо концентрація останнього збільшується, то інгібується активність одного з початкових ферментів (Е1). Така регуляція за принципом зворотного зв'язку (ретроінгібування) дозволяє контролювати вихід кінцевого продукту. Наприклад, при надлишку в клітині АТФ, яка утворюється під час гліколізу, відбувається ретроінгібування алостеричних ферментів фосфофруктокінази та піруваткінази (рис. 1.19).
Гормони також можуть виконувати роль алостеричних інгібіторів. Так, алостеричним інгібітором інсуліну є гормони надниркових залоз – глюкокортикоїди, а жіночі статеві гормони (естрогени) є алостеричними інгібіторами фермента глутаматдегідрогенази, яка каталізує дезамінування глутамінової кислоти.
Читайте також:
- Визначення опорних реакцій трьохшарнірної арки
- Визначення реакцій балки
- Визначення реакцій в’язей
- Викривлення реакцій горя
- Елементи кінетики ланцюгових реакцій
- Ембріологія ока. Вікові особливості зорових рефлекторних реакцій
- ЕНЕРГЕТИКА І КІНЕТИКА РЕАКЦІЙ.
- Закон діючих мас. Константи рівноваги гомогенних реакцій
- Замикання тимчасових зв’язків. Нейронні кореляти формування і прояву умовно-рефлекторних реакцій.
- Кінетика гетерогенних реакцій за стаціонарною лінійною дифузією
- Кінетика екстракції
- Кінетика зворотних реакцій першого порядку
| <== попередня сторінка | | | наступна сторінка ==> |
| Механізм дії ферментів | | | Застосування ферментів у медицині |
|
Не знайшли потрібну інформацію? Скористайтесь пошуком google: |
© studopedia.com.ua При використанні або копіюванні матеріалів пряме посилання на сайт обов'язкове. |