- Гідрологія і Гідрометрія
- Господарське право
- Економіка будівництва
- Економіка природокористування
- Економічна теорія
- Земельне право
- Історія України
- Кримінально виконавче право
- Медична радіологія
- Методи аналізу
- Міжнародне приватне право
- Міжнародний маркетинг
- Основи екології
- Предмет Політологія
- Соціальне страхування
- Технічні засоби організації дорожнього руху
- Товарознавство продовольчих товарів
Тлумачний словник
Авто
Автоматизація
Архітектура
Астрономія
Аудит
Біологія
Будівництво
Бухгалтерія
Винахідництво
Виробництво
Військова справа
Генетика
Географія
Геологія
Господарство
Держава
Дім
Екологія
Економетрика
Економіка
Електроніка
Журналістика та ЗМІ
Зв'язок
Іноземні мови
Інформатика
Історія
Комп'ютери
Креслення
Кулінарія
Культура
Лексикологія
Література
Логіка
Маркетинг
Математика
Машинобудування
Медицина
Менеджмент
Метали і Зварювання
Механіка
Мистецтво
Музика
Населення
Освіта
Охорона безпеки життя
Охорона Праці
Педагогіка
Політика
Право
Програмування
Промисловість
Психологія
Радіо
Регилия
Соціологія
Спорт
Стандартизація
Технології
Торгівля
Туризм
Фізика
Фізіологія
Філософія
Фінанси
Хімія
Юриспунденкция
РЕАКЦІЇ ВІДЩЕПЛЕННЯ (ЕЛІМІНУВАННЯ).
Реакціями відщеплення називаються реакції, при яких з вихідної молекули видаляються два атоми або дві групи без заміщення іншими атомами або групами. В більшості таких реакцій атоми або групи відщеплюються від двох сусідніх атомів вуглецю (причому від одного відщеплюється протон, а від другого нуклеофіл), і між цими атомами утворюється кратний зв’язок (подвійний або потрійний).
Добре відома реакція відщеплення галогеноводню, яка каталізується основами, від алкілгалогенідів: OH–
RCH2CH2Hal → RCH=CH2 + H2O + Hal
або дегідратація спиртів, яка каталізується кислотами:
+H+
RCH2CH2OH → RCH=CH2 + H3O+
При реакції β-відщеплення, атом вуглецю, від якого відходить нуклеофіл називають α-вуглецевим атомом, а вуглець, який втрачає протон – β-вуглецевим атомом.
Відомі також реакції, при яких дві групи відщеплюються від одного атома вуглецю (α-відщеплення) або від не вуглецевих атомів, як у випадку відщеплення по реакції зворотній приєднанню по карбонільній групі:
R–CH–OH → R–HC=O
| -HCN
CN
Було показано, що реакції відщеплення можуть проходити, як по моно-, так і по бімолекулярному механізму. Відповідно ці реакції позначають Е1 і Е2. При механізмі Е1, як і у випадку механізму Sn1, швидкість реакції залежить тільки від концентрації субстрату, тобто в лімітуючій стадії приймають участь тільки молекули субстрату. Так у випадку реакції за участю бромистого етилу, швидкість процесу є фактично швидкістю утворення карбкатіону, що входить до складу іонної пари.
Me3CBr → Me3C+ + Br –
За цією стадією може слідувати швидка атака іншими компонентами системи, наприклад, гідроксил-аніоном або молекулою води. Якщо при атаці реагенти діють як нуклеофіли, тоді має місце реакція заміщення. Коли ж реагенти діють як основи (тобто як донори електронної пари по відношенню до водню), в цьому випадку результатом реакції буде відщеплення протону від β-вуглецевого атому з утворенням алкену:
H Me
| | OH–
CH2–C+ → CH2=CMe2 + H2O
|
Me
Очевидно, що умови, які сприяють проходженню реакції по механізму Sn1, будуть сприяти і проходженню реакції по механізму E1, оскільки в обох випадках визначальною стадією є утворення карбонієвого катіона. Дійсно було показано, що відношення мономолекулярного відщеплення до заміщення зберігається майже постійним для даної алкільної групи, незалежно від того яка група відщеплюється у вигляді аніона. Звідси витікає, що реакції E1 і Sn1 є пов’язаними і конкуруючими, а проміжним продуктом є карбокатіон. Але зміна структури алкільної групи суттєво змінює співвідношення заміщення і відщеплення. Було показано, що розгалуження при β-атомі сприяє відщепленню по Е1. Так у випадку MeCH2CMe2Cl утворюється тільки 34% алкену, тоді як для Me2CHCMe2Cl вже 62%. Такий ефект можна пояснити стеричними причинами.
По механізму Е1 протікає також реакція дегідратації спиртів, яка каталізується кислотами:
+H+ -H2O -H+
Me3C–OH → Me3C–+OH2 → Me3C+ → Me2C=CH2
У випадку механізму Е2 швидкість відщеплення, наприклад галогеноводню від галогеналкілу, залежить від концентрації галогеналкілу і гідроксид-аніону. Цю залежність можна пояснити тим, що відщеплення протону від β-атому під дією основи відбувається одночасно з відщепленням галогенід-іона від α-атому вуглецю:
HO– + H–CH2–CH2–Cl → H2O + Н2C=CН2 + Cl–
Той факт, що лімітуючою стадією процесу є розрив зв’язку С–Н, доведено наявністю первинного кінетичного ізотопного ефекту, який спостерігається при заміні атома протія при β-вуглецю на атом дейтерія.
Було показано, що реакція відщеплення, яка протікає по механізму Е2, стереоспецифічна: вони протікають значно швидше, якщо групи, що відщеплюються, розташовані в транс-положенні одна до іншої. Така стереоспецифічність нагадує характерну для реакції Sn2 атаку "ззаду". Електронна пара, що звільнюється при видаленні протону від β-атому вуглецю, ймовірно, атакує α-вуглецевий атом "ззаду", що створює можливість відщеплення групи. В тих випадках, коли відсутність вільного обертання навколо зв’язку є перепоною сприятливій орієнтації груп, які відщеплюються, відщеплення протікає значно важче. Так було виявлено, що у випадку гексахлорциклогексану один з ізомерів відщеплює HCl на 4 порядки повільніше, чим інші ізомери, при чому цей ізомер відрізняється від інших тим, що він не має жодної пари транс-розміщених галогенів. Аналогічна ситуація спостерігається, коли групи, що відщеплюються, просторово зафіксовані подвійним зв’язком. Так відщеплення HCl протікає значно легше для хлорфумарової кислоти, чим для хлормалеїнової:
Cl–C–COOH -HCl C–COOH -HCl Cl–C–COOH

 || ||
|| ||
HOOC–C–H легко C–COOH важко H–C–COOH
Багато реакцій відщеплення, які каталізуються основами, для алкілгалогенідів і онієвих солей (+NR4, +SR3) повинні приводити до утворення різних алкенів. Співвідношення в якому фактично утворюються алкени залежить:
1) від відносної легкості відщеплення протону;
2) від доступних альтернативних β-положень;
3) від відносної стабільності перехідного стану;
4) від стеричних факторів (розміну основи і групи, що відщеплюється).
Узагальнення експериментальних даних привели до двох правил. Згідно правила Зайцева, яке справедливе для відщеплення у алкілгалогенідах і спиртах, має місце переважне утворення алкену, який несе більшу кількість алкільних груп. Правило Зайцева пояснюється 3 фактором, тобто вищою стабільністю вторинного карбокатіона. Згідно правила Гофмана, яке справедливе більше для онієвих сполук, має місце переважне утворення менш стабільного алкену, який несе меншу кількість алкільних груп (тобто α-олефінів). Так при нагріванні
 Me MeCH2CH2NMe2 + CH2=CH2
Me MeCH2CH2NMe2 + CH2=CH2
|
 Me–CH2–CH2–N+–CH2–CH3
Me–CH2–CH2–N+–CH2–CH3
|
Me MeCH=CH2 + Me2NCH2CH3
утворюється головним чином етилен, а не пропілен. Це можна пояснити виходячи з 1 фактору. Індуктивний ефект метильної групи в пропільному заміснику понижує здатність до відщеплення атомів водню, які зв’язані з β-атомом вуглецю цього замісника. В етильному заміснику подібний ефект відсутній і доцільнішим є відрив протону від β-вуглецевого атома етильного замісника. Таким чином можна вважати, що при відщепленні по Зайцеву найбільш важливим є ефекти спряження алкільних груп, тоді як при відщепленні по Гофману переважають індуктивні ефекти. Збільшення ролі стеричних факторів (розміру групи, що відривається, і розгалуженості вихідної сполуки) приводить до збільшення ролі відщеплення по Гофману.
Реакції α-відщеплення зустрічаються рідше. Відомим прикладом такої реакції є гідроліз хлороформу під дією сильної основи:
швидко повільно швидко повільно
HO– + HCCl3 → –CCl3 → Cl– + :CCl2 → CO → HCOO–
-H2O +H2O OH–
Початкова атака гідроксид-аніоном водню, а не вуглецю, зумовлена електроноакцепторною дією атомів хлору, яка підвищує кислотність водню. Рухливість водню підтверджується легкістю ізотопного обміну водню на дейтерій.
КАРБАНІОНИ І ЇХ РЕАКЦІЇ.
В органічній хімії відомі сполуки, які функціонують як кислоти в класичному розумінні. Вони розщеплюються з відривом протону від зв’язку C–H, з утворенням спряженої основи, яку називають карбоаніоном.
H3C–H + :B ↔ H3C–– + HB+
В даному випадку має місце не швидкий відрив протону від О–Н зв’язку, а відносно повільний відрив від С–Н зв’язку. Не дивно, що у випадку відриву протону від аліфатичних сполук, така здатність проявляється слабо. Це зумовлено міцністю таких зв’язків і для них малохарактерна здатність до стабілізації карбоаніону, що утворився. Тому про кислотність метану можна говорити тільки умовно, рК≈43. Введення сильних електроноакцепторних груп збільшує рухливість атома водню і, як наслідок триціанметан по силі кислотності близький до мінеральних кислот. Карбоаніони, які неможливо отримати прямим відривом протону основою, часто можуть бути отримані дією металів на відповідні галогеніди
R–Br + 2M → R–M + M–Br
Чим менш електровід’ємний метал, тим більш полярним буде зв’язок R–M і сильніше повинен бути виражений карбоаніонний характер R.
Карбокатіони можуть бути стабілізовані за рахунок збільшення вкладу s-електронів в гібридизовану орбіталь, яка виникає при втраті протону. Якщо карбоаніони містять достатньо сильні електроноакцепторні групи або атоми, то вони можуть бути стабілізовані за рахунок тільки індуктивних ефектів. Реально такий ефект спостерігається для четвертинного азоту і фтору. Електроноакцепторні групи, які мають чіткий мезомерний ефект більш суттєво впливають на стабілізацію карбоаніону. Так карбонільна і нітрогруппа виявляються більш ефективними стабілізуючими замісниками ніж четвертинний азот і трифторметильна група.
Викликає інтерес факт високої стабільності аніона циклопентадієнілу, яка зумовлена наявністю шести π-електронів системи, які утворюють квазіароматичну структуру.
Для карбоаніонів, як і для карбокатіонів, характерним є явище таутомерних перетворень. Так кето-енольні перетворення протікають з участю карбоаніонів:
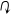 |
B: H R' OH
| | |
R–C–C=O R–C=C–R'
 | |
| |
 |  |
 R––C–C=O R–C=C–O–
R––C–C=O R–C=C–O–
| | | |
R' R'
 В даному випадку таутомерне перетворення легше протікає по ступінчатому механізму, так як в проміжній стадії має місце утворення досить стабільного карбоаніону. Таутомерні перетворення можуть протікати і по синхронному механізму. При синхронному механізму процеси дейтерообміну і таутомерного перетворення повинні протікати з одною і тією ж швидкістю, при чому дейтерообмін неможливий без таутомерії.
В даному випадку таутомерне перетворення легше протікає по ступінчатому механізму, так як в проміжній стадії має місце утворення досить стабільного карбоаніону. Таутомерні перетворення можуть протікати і по синхронному механізму. При синхронному механізму процеси дейтерообміну і таутомерного перетворення повинні протікати з одною і тією ж швидкістю, при чому дейтерообмін неможливий без таутомерії.
 У випадку ступінчатого механізму дейтерообмін може проходити і без таутомерного перетворення за рахунок зворотньої реакції.
У випадку ступінчатого механізму дейтерообмін може проходити і без таутомерного перетворення за рахунок зворотньої реакції.
Таким чином швидкість дейтерообміну повинна бути рівна або більша швидкості таутомерного перетворення. Вимірювання показали, що обидва ці процеси протікають з приблизно однаковою швидкістю і, якщо відомо, що реакція зворотня, можна вважати, що вона протікає по синхронному механізму.
Розрив С–Н зв’язку відбувається з певною швидкістю, яку можна оцінити по швидкості дейтерообміну. Приблизно можна вважати, що швидкість заміщення дейтерієм водню рівна швидкості утворення проміжного аніону (при подачі основи), а при наступному підкисленні – швидкості утворення таутомерів з цього аніону.
Таутомерні перетворення найбільш повно вивчені для випадку кето-енольної таутомерії. Так вміст енольної форми для ацетону незначний, тоді як β-дікетони існують переважно в енольній формі. Енольна форма може стабілізуватися наявністю одного або кількох замісників, які здатні її стабілізувати за рахунок делокалізації π-електронів подвійного вуглець-вуглецевого зв’язку:
O=C–C=C–OH → –O–C=C–C=+OH
| | | | | |
Додатковий фактор стабілізації енольної форми є можливість утворення внутрішнього водневого зв’язку, наприклад:

Утворення водневого зв’язку приводить до зменшення полярності енолу і підвищення його компактності і, як наслідок, зниження температури кипіння. Вміст кето- і енольної форм залежить від полярності розчинника. Так для ацетилацетону в гексані вміст енольної форми досягає 92%, тоді як у воді (де кетоформа може утворювати водневі зв’язки з молекулами води) переважає кетоформа – 85%.
За участю карбоаніонів протікає значна кількість реакцій ПРИЄДНАННЯ: приєднання реактивів Гриньяру і ацетиленід-іону до карбонільної групи, альдольна конденсація, конденсація Кляйзера і т.д. Алкільні і арильні похідні, ацетиленіди металів більш позитивних ніж магній, реактиви Гриньяра здатні приєднувати дуже слабкий електрофільний двохокис вуглецю з утворенням відповідної карбонової кислоти.
M+R– + CO2 → R–COO–M+
Реакцію звичайно проводять придаючи розчин металоорганічних сполук в інертному розчиннику до великого надлишку дрібнодисперсного твердого двохокису вуглецю; особливо широко ця реакція використовується для отримання кислот ацетиленового ряду.
Карбоаніони приймають участь і у великій кількості реакцій ЗАМІЩЕННЯ. Карбоаніони, які утворюються з малонового ефіру, β-кетоефірів, β-дікарбонільних сполук, легко вступають в реакції заміщення з алкілгалогенідами. Ця реакція слугує хорошим методом утворення нових вуглець-вуглецевих зв’язків.
В більшості випадків карбаніон утворюється в неводних розчинах в присутності основ, таких як EtO–, і далі реакція протікає по звичайному механізму Sn2.
EtO– Br–
(EtOOC)2HC–H → (EtOOC)2HC– + H3–C–Br → (EtOOC)2HC••CН3••Br → (EtOOC)2HC–CН3
В деяких випадках механізм Sn2 був підтверджений кінетичними методами, було показано обертання конфігурації при активному атомі вуглецю.
Вважається, що у розчинниках РЕАКЦІЯ ВЮРЦА теж протікає через проміжне утворення карбоаніона, який далі реагує з другою молекулою алкілгалогеніду з утворенням змішаного вуглеводню з досить високим виходом:
2Na _ +R′Cl
R–CH2CH2Cl → NaCl + RCH2CH2Na → RCH2CH2R′
Додаткові дані на користь іонного, а не радикального, механізму реакції Вюрца було отримано при вивченні оптично активних алкілхлоридів. Було показано, що атака другою молекулою алкілхлориду карбоаніоном проходить по механізму Sn2 і приводить до обернення конфігурації по атомі вуглецю.
РЕАКТИВ ГРИНЬЯРА можна розглядати як джерело карбоаніонів. Було показано, що натрієві і калієві похідні реагують у формі істинних карбоаніонів. Вклад ковалентного зв’язку у реактиві Гриньяра значно вагоміший. Так альдегіди отримують при взаємодії реактиву Гриньяра з ортомурашиним ефіром.
H+, H2O

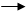 R––Mg+Br + CH(OEt)3 R–CH(OEt)2 R–CHO
R––Mg+Br + CH(OEt)3 R–CH(OEt)2 R–CHO
Прикладом карбоаніона може слугувати фенолят-аніон, який утворюється внаслідок делокалізації π-електронів з переносом від’ємного заряду з кисню на вуглець:

В реакції Кольбе-Шмідта фенолят-аніон атакує молекула двохокису вуглецю з утворенням саліцилової кислоти:

При цьому утворюються тільки сліди натрієвої солі п-оксибензойної кислоти, але коли використовується фенолят калію основним продуктом реакції є сіль п-кислоти. Прийнято вважати, гідроксид натрію сприяє атаці в орто-положення за рахунок утворення хелатної сполуки у вигляді іонної пари.
Іон калію, розмір якого більше, напевно мало ефективний у цій ролі.
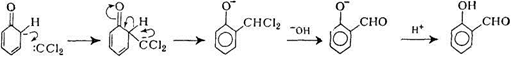 В реакції Реймера-Тімана фенолят-аніон взаємодіє з хлороформом в присутності сильної основи, з утворенням переважно орто-саліцилового альдегіду, кількість пара-ізомеру незначна:
В реакції Реймера-Тімана фенолят-аніон взаємодіє з хлороформом в присутності сильної основи, з утворенням переважно орто-саліцилового альдегіду, кількість пара-ізомеру незначна:
Фенолят-аніон атакує електронодефіцитну молекулу карбена: CCl2, яка утворюється при гідролізі хлороформу в лужному середовищі. Дихлорметильне похідне в умовах реакції гідролізується до альдегіду.
Добре вивчено реакцію ГАЛОГЕНУВАННЯ КЕТОНІВ, яка протікає через проміжне утворення карбоаніона. Швидкість реакції бромування ацетону залежить від концентрації ацетону та основи і не залежить від концентрації брому. Було зроблено висновок, що лімітуючою стадією реакції є утворення карбоаніону при взаємодії ацетону з гідроксилом, за якою наступає швидке бромування:
OH– _ Br2
Me–CО–Me ↔ [ Me–CО–CH2 → Me–CО–=CH2 ] → Me–CО–CH2Br
Було показано, що кетони хлоруються, йодуються, обмінюють дейтерій зі швидкістю, яка рівна швидкості бромування, що підтверджує вказаний механізм реакції. Атоми водню при вуглецю, до якого приєднано галоген, стають більш кислі, легше вступають в подальшу реакцію. Додатковим фактором є стабілізація карбоаніона за рахунок від’ємного індукційного ефекту галогена. OH– _
CH3–CО–CH2–Br ↔ CH3–CО–CH–Br → CH3–CО–CHBr2
Приєднання третього атому галогена проходить ще швидше. Реакція закінчується атакою гідроксид-аніоном карбонільного вуглецю, який має чітко виражений позитивний заряд.
Me–CО–+CBr3 + ОН– → Me–CООН + –CBr3 → [ Me–CОО ]+ + HCBr3
При використанні несиметричних кетонів, має місце атака атому вуглецю з менш електронодонорними властивостями, тобто у випадку етилметилкетону по метильній групі.
Інша картина спостерігається при кислому каталізі галогенування кетонів. Ймовірно реакція протікає через енольну форму кетону, утворення якої і є лімітуючою стадією реакції:
O OH O
_ || | ||
A + H–CH2–C–CH3 + H+ ↔ CH2=CH–Me + Br2 → BrCH2–C–CH3
В даному випадку вплив замісника протилежний попередньому. Більш стабільною енольною формою за рахунок зверхспряження буде етильна група і галогенування пройде переважно по α-вуглецю довшого ланцюга. Від’ємний індукційний ефект галогену стабілізує кето-форму. Тому бромування ацетону проходить легше ніж монобромпохідного. Це дає можливість виділити монобромпохідне. Надалі реакція проходить по метильній, а не по метиленовій групі.
Ще одним типом реакцій за участю карбоаніонів є реакція ДЕКАРБОКСИЛЮВАННЯ. Аніони карбонових кислот декарбоксилюються з утворенням карбоаніона, який приєднує протон з розчинника або іншого джерела:
_ _ H+
O–CO–R → O=C=O + R → R–H + CO2
Введення електроноакцепторних груп в радикал R повинно стабілізувати карбоаніон і, як наслідок, збільшувати швидкість реакції. Дійсно аніон нітрооцтової кислоти розкладається з дуже високою швидкістю.
_ _ _
O–C–CH2–NO2 → [CH2–N+=O → CH2=N+–O ] → CH3NO2
|| -CO2 | |
O O– O–
Утворення проміжного карбоаніона підтверджується добавкою брому, коли в продуктах реакції присутній бромнітрометан, хоча добре відомо, що нітрооцтова кислота і нітрометан в реакцію з бромом не вступають. Напрошується висновок, що проходить бромування карбоаніона, який утворюється після декарбоксилювання.
Аналогічно протікає декарбоксилювання b-кетокислот:
_ _
O–C–CH2–C–Me → [CH2–C–Me → CH2=C–Me ] → CH3COMe
|| || -CO2 || |
O O O O–
Знайдено, що швидкість реакції декарбоксилювання залежить від концентрації кетокислоти і її аніону. Це зумовлено тим, що перенос протону до кетогрупи починається з утворенням водневого зв’язку:
O OH O–H O
|| | | ||
Me–C–CH2–C=O → Me–C=CH2 + C=O → CH3COMe + CO2
Перегрупування карбоаніонів зустрічаються рідко. Прикладом перегрупування карбоаніона може слугувати реакція переміщення арильної групи:
Na _
Ph3C–CH2Cl → [Ph3C–CH2 +Na ] _
COO Na+
_ CO2 |
Ph2CH–CH2Ph → Ph2C–CH2Ph + Na+ → Ph2C–CH2Ph
Те, що реакція протікає через утворення карбоаніона, свідчить про те, що її продукти, мають ту ж будову як і продукти, які повинні утворитися в результаті протонування і карбоксилювання карбоаніона. Якщо замість натрію використовувати літій і проводити реакцію при пониженій температурі, реакція перегрупування не проходить. Але при нагріванні реакція протікає легко. Більш жорсткі умови проведення реакції перегрупування для літійпохідних пов’язують з більш ковалентним зв’язком C–Li ніж C–Na. Переміщення алільної групи нехарактерне для карбоаніонів.
КАРБЕНИ.
Карбени високореакційноздатні частинки; практично у всіх карбенів період напівперетворення значно менший 1с, їх вдається отримати тільки при низьких температурах. Родоначальний карбен CH2 називають звичайно метиленом, СCl2 – дихлорокарбеном.
Найбільш поширеними методами отримання карбенів є:
1. При α-відщепленні від вуглецю спочатку відщеплюється група без електронної пари, звичайно протон, а потім група з електронною парою, звичайно галоген-іон
H
| -H+ _ -Cl– _
R–C–Cl → R–C–Cl– → R–C
| | |
R R R
Типовим прикладом служить утворення дихлоркарбена при обробці хлороформу основами. З хорошими виходами утворюються карбени по реакціях:
CCl3COO– → CCl2 + CO2 + Cl–
NaI
Ph–Hg–CF3 → CF2 + PhHgI + NaF
2. Розпад сполук, які містять певні типи подвійних зв’язків:
_ _
R2C=Z → R2C + Z
Найбільш важливим методом отримання метилену є фотоліз кетену
_ _
CH2=C=O → CH2 + C≡O+
і ізоелектронний розклад діазометану
_
CH2=N=N → CH2 + N2
Ізомерні діазоалканам діазіріни також утворюють карбени
N
/ _
R2C → R2C + N2
\
N
Через високу реакційну здатність карбенів часто досить складно довести, що вони дійсно приймають участь в реакції. Часто немає ніяких прямих доказів участі карбенів в реакції. Реакції карбенів достатньо різноманітні.
1. Приєднання до подвійних вуглець-вуглецевих зв’язків. При взаємодії карбенів з цис-2-бутеном утворюється циклопропан:
H H H CH2 H
_ \ / \ / \ /
CH2 + C=C → C––C
/ \ / \
Me Me Me Me
Карбени також приєднуються до ароматичних систем, але продукти приєднання зразу ж перегруповуються, звичайно з розширенням циклу.
2. Впровадження карбенів по зв’язку C–H. Так при взаємодії карбену з метаном утворюється етан, а при взаємодії з пропаном – н-бутан і ізобутан. Хоча ці реакції не представляють інтерес з точки зору синтезу, вони показують виняткову реакційну здатність карбенів. Реакція з вищими алканами проходить низькоселективно.
3. Висока реакційна здатність карбенів, малий час їх життя, відповідно низька їх концентрація, практично не приводить до їх реакції димеризації з утворенням алкенів.
4. Алкілкарбени легко перегруповуються з міграцією алкілу або водню. Такі реакції настільки швидкі, що реакція впровадження або приєднання по подвійному зв’язку зустрічається рідко. Перегрупування відразу приводить до утворення стійких молекул:
_
CH3–CH2CH2–CH → CH3–CH2–CH=CH2
_
R–CО–CH → O=C=CH–R
Перетворення арилкарбенів в кетени відоме як перегрупування Вольфа.
РАДИКАЛИ І ЇХ РЕАКЦІЇ.
Поряд з гетерогенним розривом зв’язків, який приводить до утворення карбокатіонів і карбаніонів, можливий і гомогенний розрив зв’язків, який приводить до утворення продуктів з неспареними електронами (радикалів). Енергія активації такого гомолізу тільки незначно перевищує енергію зв’язку, який розривається. Тому швидкість гомолітичного розриву повинна збільшуватись зі зменшенням енергії зв’язку.
Радикальні реакції часто протікають в газовій фазі, але можуть проходити і в розчинах особливо малополярних розчинників. Каталізаторами радикальних реакцій є світло і речовини, що легко утворюють радикали. Після первинного утворення радикалу наступна стадія протікає швидко з низькою селективністю. Для радикальних реакцій є характерним ланцюговий характер, коли первинний радикал при взаємодії з нейтральною молекулою викликає утворення нового радикалу. Після того як радикальна реакція почалась, надалі вона протікає з дуже великою швидкістю внаслідок розвитку швидких ланцюгових реакцій.
Основними рисами радикальних реакцій є висока швидкість, здатність ініціюватися радикалами або їх ініціаторами, здатність сповільнюватися в присутності інгібіторів. Якщо є необхідність підтвердити радикальний механізм реакції, найпростішим методом є спостереження за зміною швидкості реакції при придачі ініціаторів радикалів (наприклад, перекисей) або інгібіторів (наприклад, гідрохінону).
Як правило радикали короткоживучі часточки, але відомий ряд стабільних радикалів, які здатні існувати на протязі тривалого часу. До стабільних можна віднести радикали на основі трифенілметану. Його стабільність зумовлена делокалізацією неспареного електрона по π-орбіталям бензольних кілець:

Дещо менш стабільні радикали, які утворюються при нагріванні тетраарилгідразинів в неполярних розчинниках, причому розчин забарвлений в зелений колір:
KMnO4

 2Ph2NH Ph2N:NPh2 2Ph2N•
2Ph2NH Ph2N:NPh2 2Ph2N•
Відносно низька енергія зв’язку N–N та ефект делокалізації сприяє дисоціації тетрафенілгідразинів.
Радикали, які живуть недовго, зустрічаються значно частіше і значно важливіші, так як за їх участю протікає велика кількість хімічних реакцій. Короткий час життя викликаний високою реакційною здатністю. Відносна стабільність простих алкільних радикалів проявляється в легкості гомолітичного розпаду відповідних вуглеводнів. По стабільності їх можна розташувати в ряд:
R3C• > R2CH• > RCH2• > CH3•
який співпадає з рядом стабільності карбокатіонів. Цей ряд відображає зменшення стабілізації за рахунок зверхспряження і зняття стеричних затруднень. Делокалізація неспареного електрона за участю π-орбіталей подвійного зв’язку приводить до зростання стабільності радикалів. Алільний і бензильний радикали достатньо стабільні:
CH2=CH–C•H2 → •CH2–CH=CH2
Радикали утворюються при фотохімічному і термічному розчепленні зв’язків, окисно-відновних реакціях з переносом одного електрону (викликаються неорганічними іонами), електролізі.
Фотохімічне розщеплення має місце при опроміненні реакційної маси сонячним світлом або під дією ультрафіолетового опромінення. Добре відомі приклади розкладу ацетону в газовій фазі під дією світла з довжиною хвилі 3000А.
Me–CО–Me → Me• + •CО–Me → 2Me• + CO
Іншим класичним прикладом є розклад молекулярного хлору в атомарний на сонячному світлі:
Cl2 → 2Cl•
який є першою стадією хлорування при освітленні. Реакції такого типу протікають не тільки в газовій фазі, але і в розчинах. При цьому слід пам’ятати, що стабільність радикалів в розчинах значно менша, що пов’язано з їх взаємодією із молекулами розчинника.
Аналогічно дії жорсткого опромінення на молекулу органічної речовини впливає і підвищена температура. Так термічний розклад вуглець-вуглецевих зв’язків, який протікає при крекінгу вуглеводнів при 600 С, приводить до утворення радикалів. Цьому процесу сприяють ініціатори, які відривають атом водню від метиленової групи, а радикал що утворюється дає олефін меншої молекулярної маси і новий радикал, який продовжує ланцюгову реакцію:
R′• + R–CH2–CH2–R → R′–H + R–•CH–CH2–R → R–CH=CH + •R
Сповільнення реакції спостерігається тоді, коли концентрація молекул з довгим ланцюгом достатньо понижається.
При температурі 300 С розкладається азометан з утворенням стабільної молекули азоту:
CH3–N=N–CH3 → 2CH3• + N2
Особливо легко розкладаються органічні перекиси, які містять слабкий зв’язок O–O. Тому перекисні сполуки використовують як ініціатори радикальних процесів:
O O O
|| || ||
Ph–C–OO–C–Ph → 2Ph–CO•
Радикали можуть приймати участь в реакціях приєднання, заміщення і в перегрупуваннях, при чому після перегрупування проходить приєднання або заміщення.
В алканах під дією майже всіх радикалів в першу чергу відривається третинний атом водню, потім вторинний. При обробці хлором або бромом субстратів, молекули яких містять подвійний зв’язок, звичайно має місце не заміщення, а відщеплення. Практично ніколи не спостерігається відрив вінільних атомів водню, тоді як алільні атоми водню відриваються набагато легше ніж любі інші водні. Атака бокового ланцюга жирноароматичних сполук переважно проходить в α-положенню відносно кільця. Це положення піддається атаці швидше ніж первинний атом водню. Ароматичні атоми водню відриваються рідко, якщо в молекулі є аліфатичні атоми водню. При галогенуванні електроноакцепторні групи суттєво дезактивують сусіднє положення. Такі сполуки, як оцтова кислота і ацетилхлорид, взагалі не піддаються атаці. Відрив галогена зустрічається значно рідше, відомо, що здатність галогенів до відриву зменшується від йоду до фтору.
Вільнорадикальне ЗАМІЩЕННЯ при ароматичному атомі вуглецю супроводжується утворенням арильного радикала.
Всі замісники (електронодонорні і електроноакцепторні) підвищують реакційну здатність в орто- і пара-положеннях по відношенню до незаміщеного бензолу. Реакційна здатність мета-положення не змінюється при введенні замісників. Реакційна здатність орто-положення дещо більша ніж пара-положення (за виключенням об’ємних замісників). Вплив замісників значно менший ніж у реакціях електрофільного і нуклеофільного заміщення: фактори парціальних швидкостей незначні.
Селективність реакцій вільнорадикального заміщення зростає зі збільшенням стабільності вільного радикалу. Так атом брому більш селективний ніж атом хлору. І якщо в субстраті є тільки первинні атоми водню, тоді реакція протікає дуже повільно або взагалі не проходить. В той же час ізобутан можна селективно бромувати до третбутилброміду з високим виходом. Але бром реагує з толуолом практично моментально.
Розчинник звичайно мало впливає на реакції вільнорадикального заміщення. Але у тих випадках, коли радикал може утворювати комплекси з молекулами розчинника, роль останнього зростає. Утворення комплексів приводить до деякої стабілізації радикалу, зменшенні його реакційної здатності і, як наслідок, збільшенні селективності.
В реакціях заміщення біля атому вуглецю, які протікають по вільнорадикальному механізму, пряме заміщення в дійсності не проходить. Насправді має місце двохстадійний процес:
Cl2 → 2Cl•
Cl• + R–H → R• + HCl
R• + Cl2 → R–Cl + Cl•
Дану реакцію можна ініціювати і підвищенням температури, але температура повинна бути суттєвою. Так при 120 С в темряві реакція практично не іде. Зі збільшенням температури швидкість реакції зростає, але слід пам’ятати, що зі збільшенням температури зростає швидкість реакції і дегідрогалогенування. Так при хлоруванні пропілену має місце утворення хлористого алілу, а не приєднання по подвійному зв’язку:
Cl2 + CH3–CH=CH2 → Cl–CH2–CH=CH2 + HCl
Селективність реакції вільнорадикального хлорування досить низька, тому для препаративних цілей вона використовується рідко. Так при хлоруванні ізобутану переважно йде заміщення третинного атому водню, але наявність дев’яти первинних атомів вирівнює співвідношення напрямку заміщення.
Реакція фторування проходить легше хлорування, вона може проходити при відсутності світла і ініціаторів. Рушійною силою реакції фторування є більш висока термодинамічна стабільність її продуктів – енергія зв’язку H–F на 100 ккал/моль перевищує енергію зв’язку F–F. Реакція фторування часто супроводжується деструкцією молекули. Для м’якого фторування доцільно використовувати фторокситрифторметан (CF3OF) при опроміненні. При відсутності опромінення CF3OF м’яко і селективно фторує третинні положення. Реакція бромування протікає значно повільніше і більш селективно, вимагає температури вище кімнатної. Пряме радикальне йодування проходить при активуючому опроміненні УФ-світлом з довжиною хвилі 184,9 нм, але реакції йодування проводять рідко, так як HI відновлює алкілйодид.
 Суттєву роль в препаративному синтезі відіграє галогенування не самими галогенами, а реактивами, які містять рухомий галоген. М’яко і з високою селективністю проходить хлорування трет-BuOCl в присутності ініціатора. З хорошим успіхом проходить хлорування під дією хлористого тіонілу в присутності ініціатора. В цьому випадку утворюється радикал •SOCl2, який легко втрачає SO2 і дає Cl•. Хорошим бромуючим агентом є N-бромсукцинамід, який селективно реагує з α-положенням по відношенню до подвійного зв’язку.
Суттєву роль в препаративному синтезі відіграє галогенування не самими галогенами, а реактивами, які містять рухомий галоген. М’яко і з високою селективністю проходить хлорування трет-BuOCl в присутності ініціатора. З хорошим успіхом проходить хлорування під дією хлористого тіонілу в присутності ініціатора. В цьому випадку утворюється радикал •SOCl2, який легко втрачає SO2 і дає Cl•. Хорошим бромуючим агентом є N-бромсукцинамід, який селективно реагує з α-положенням по відношенню до подвійного зв’язку.
Іншим прикладом реакції вільнорадикального заміщення є автоокислення – реакція органічних сполук з киснем в м’яких умовах. Розклад більшості органічних речовин на повітрі під дією світла зумовлений фотоокисленням, таким шляхом руйнується каучук і більшість пластмас, тверднуть лаки і фарби. Ініціаторами таких процесів є важкі метали та перекиси. Молекула кисню, яка має бірадикальну природу, активно реагує з радикалами:
–C• + •O–O• → –C–OO•
–C–OO• + –C–H → –COOH + –C•
В багатьох випадках гідроперекис, що утворився, може виступати в ролі ініціатора, і тоді реакція стає автокаталітичною.
Ще одним прикладом вільнорадикального заміщення є реакція Зандмейєра – взаємодія солей діазонію з хлористою міддю. Вважається, що в цій реакції арильний радикал утворюється по реакції:
ArN2+ + CuCl2– → Ar• + N2 + CuCl2
Ar• + CuCl2 → Ar–Cl + CuCl2
Аналогічний процес, що проводять з міддю в присутності HCl або HBr називають реакцією Гатермана. Арилхлориди і арилброміди отримують по таких реакціях з високим виходом.
Арильний радикал, що утворюється з бензоліазонію, може вступати в широке коло реакцій. Діазогрупа може бути заміщена на водень, нітрогрупу, ціангрупу (реакція Зандмейєра), іншу арильну групу (реакція Гомберга) і т.д.
Важливою групою радикальних реакцій є реакції ПРИЄДНАННЯ. Приєднання галогенів може проходити як по іонному, так і по радикальному механізму. В газовій фазі при освітленні має місце виключно радикальне приєднання. Те ж відноситься і до розчинів в неполярних розчинниках при освітленні. В полярних розчинниках без освітлення, особливо в присутності кислот Льюїса, приєднання проходить виключно по іонному механізму. Фотохімічне приєднання хлору до тетрахлоретилену можна представити схемою:
h
Cl–Cl → 2Cl•
CCl2=CCl2 + •Cl → •CCl2–CCl3
•CCl2–CCl3 + Cl2 → CCl3–CCl3 + Cl• і т.д.
Початковою стадією реакції є фотохімічне розщеплення хлору з утворенням двох атомів – радикалів – хлору. Кожний атом хлору ініціює швидку ланцюгову реакцію. До останніх стадій реакції, поки не прореагували всі ненасичені сполуки, концентрація Cl• буде дуже низькою. Тому ймовірність рекомбінації радикалів буде незначна, і реакція рекомбінації проходить рідко. Реакція інгібується киснем, як бірадикальною часточкою. Кінцева стадія, тобто атака радикалом хлору, екзотермічна. Тому приєднання хлору проходить швидко і характеризується довгими реакційними ланцюгами. Приєднання брому проходить повільніше, реакційні ланцюги коротші. Приєднання брому при звичайних умовах взагалі не відбувається.
Приєднання хлору до бензолу – одна з небагатьох реакцій приєднання по неактивованому бензольному кільці – також проходить по радикальному механізму; показано, що реакція каталізується світлом і перекисами і сповільнюється класичними інгібіторами.
Реакція взаємодії бромистого водню і пропілену в присутності ініціаторів радикалів проходить швидко по ланцюговому механізму з утворенням 1-бромпропану (тобто всупереч правилу Марковнікова). Причина такого приєднання полягає в тому, що серед можливих первинних і вторинних радикалів другі більш стабільні і вони визначають характер другого приєднання.
Ймовірність протікання реакції приєднання по іонному або радикальному механізму визначається наявністю домішок, які можуть виступати як ініціатори або інгібітори. Таким чином придача певних речовин може забезпечити повний контроль за селективністю реакції.
Серед вільнорадикальних реакцій вінільна полімеризація в промисловості використовується найбільш часто. Вона протікає в три стадії:
– утворення радикалів з ініціаторів;
– ріст ланцюга: CH2=CH2
 R• + CH2=CH2 RCH2–CH2• R–(CH2–CH2)2 і т.д.
R• + CH2=CH2 RCH2–CH2• R–(CH2–CH2)2 і т.д.
– обрив ланцюга:
R–(CH2–CH2)n–CH2–CH2• + R• R–(CH2–CH2)n+1–R
Ріст ланцюга проходить звичайно з високою швидкістю. Кисень повітря виступає ініціатором реакції, тому до мономера звичайно добавляють інгібітори. Якщо стабілізований мономер необхідно включити в реакцію тоді добавляють таку кількість ініціатора, яка б зв’язала дію інгібітора і почала ланцюгову реакцію.
Ініціатори не можна розглядати як каталізатори реакції, так як вони в кінцевому варіанті приєднуються до продуктів полімеризації і не регенеруються. Довжину ланцюга і швидкість реакції полімеризації можна регулювати кількістю завантаженого ініціатора.
Читайте також:
- VIII. Реакції, в результаті яких утворюються високомолекулярні сполуки
- АДАПТАЦІЙНІ РЕАКЦІЇ М'ЯЗОВОЇ СИСТЕМИ
- АДАПТИВНІ РЕАКЦІЇ МІКРООРГАНІЗМІВ НА СТРЕСОВІ ДІЇ.
- Аналітичні реакції та вимоги, яким вони повинні відповідати.
- Антигени. Антитіла. Серологічні реакції .
- Біогенні аміни та реакції декарбоксилювання
- Види опор та їх реакції
- Визначення реакції продуктів, кислотності і лужності.
- В’язі. Реакції в’язей.
- Гетерогенні реакції.
- Гомогенні реакції.
- Дегідратація спиртів (відщеплення води).
| <== попередня сторінка | | | наступна сторінка ==> |
| РЕАКЦІЯ МІХАЕЛЯ. | | | Навчання персоналу |
|
Не знайшли потрібну інформацію? Скористайтесь пошуком google: |
© studopedia.com.ua При використанні або копіюванні матеріалів пряме посилання на сайт обов'язкове. |