РЕЗОЛЮЦІЯ: Громадського обговорення навчальної програми статевого виховання
ЧОМУ ФОНД ОЛЕНИ ПІНЧУК І МОЗ УКРАЇНИ ПРОПАГУЮТЬ "СЕКСУАЛЬНІ УРОКИ"
ЕКЗИСТЕНЦІЙНО-ПСИХОЛОГІЧНІ ОСНОВИ ПОРУШЕННЯ СТАТЕВОЇ ІДЕНТИЧНОСТІ ПІДЛІТКІВ
Батьківський, громадянський рух в Україні закликає МОН зупинити тотальну сексуалізацію дітей і підлітків
Відкрите звернення Міністру освіти й науки України - Гриневич Лілії Михайлівні
Представництво українського жіноцтва в ООН: низький рівень культури спілкування в соціальних мережах
Гендерна антидискримінаційна експертиза може зробити нас моральними рабами
ЛІВИЙ МАРКСИЗМ У НОВИХ ПІДРУЧНИКАХ ДЛЯ ШКОЛЯРІВ
ВІДКРИТА ЗАЯВА на підтримку позиції Ганни Турчинової та права кожної людини на свободу думки, світогляду та вираження поглядів
- Гідрологія і Гідрометрія
- Господарське право
- Економіка будівництва
- Економіка природокористування
- Економічна теорія
- Земельне право
- Історія України
- Кримінально виконавче право
- Медична радіологія
- Методи аналізу
- Міжнародне приватне право
- Міжнародний маркетинг
- Основи екології
- Предмет Політологія
- Соціальне страхування
- Технічні засоби організації дорожнього руху
- Товарознавство продовольчих товарів
Тлумачний словник
Авто
Автоматизація
Архітектура
Астрономія
Аудит
Біологія
Будівництво
Бухгалтерія
Винахідництво
Виробництво
Військова справа
Генетика
Географія
Геологія
Господарство
Держава
Дім
Екологія
Економетрика
Економіка
Електроніка
Журналістика та ЗМІ
Зв'язок
Іноземні мови
Інформатика
Історія
Комп'ютери
Креслення
Кулінарія
Культура
Лексикологія
Література
Логіка
Маркетинг
Математика
Машинобудування
Медицина
Менеджмент
Метали і Зварювання
Механіка
Мистецтво
Музика
Населення
Освіта
Охорона безпеки життя
Охорона Праці
Педагогіка
Політика
Право
Програмування
Промисловість
Психологія
Радіо
Регилия
Соціологія
Спорт
Стандартизація
Технології
Торгівля
Туризм
Фізика
Фізіологія
Філософія
Фінанси
Хімія
Юриспунденкция
Організація та алгоритм діагностики ФКУ
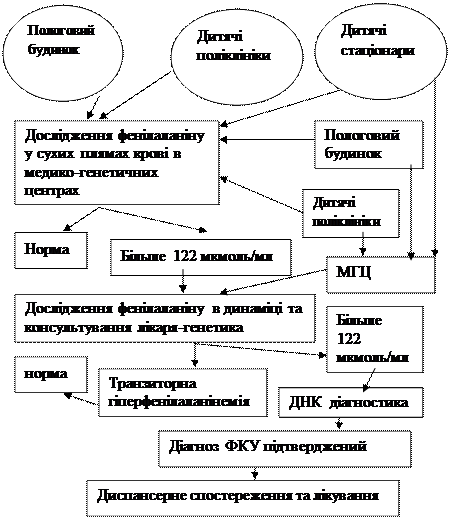 |
Діагноз ФКУ виставляється на основі клінічної картини і результатів біохімічного обстеження сечі (фенілпіровиноградна кислота) чи крові (фенілаланінемія).
Програма диспансерного спостереження включає регулярний контроль рівня ФА в крові, систематичну оцінку соматичного, неврологічного та психічного статусу, ультразвукове спостереження за станом внутрішніх органів, лабораторний контроль показників крові, ДНК-діагностику для визначення характеру мутації, електрофізіологічні методи дослідження тощо.
Схема обстеження дитини, хворої на ФКУ:
| Захід | Вік дитини | |||
| до 1-го року | 1-3 роки | 3-5 років | Більше 5 років | |
| Консультація генетика | Кожні 3 місяці | Кожних 4 місяці | Кожних 6 місяців | 1 раз на рік |
| Консультація дієтолога | 2 рази на тиждень | 2 рази на тиждень | 2 рази на місяць | 1 раз на рік |
| Аналіз крові на ФА | 1 раз на два тижні | 2 рази на місяць | 1 раз на місяць | 1 раз на 6 місяців |
| Консультація психолога | Кожних 3 місяці | Кожних 3 місяці | Щороку | Щороку |
| Клінічний аналіз крові, клінічний аналіз сечі | 1 раз на півроку | 1 раз на півроку | Щороку | Щороку |
| Біохімічний аналіз крові з дослідженням функції печінки, нирок | Щороку | |||
| Електроенцефалограма | Щороку | |||
| Ультразвукове дослідження внутрішніх органів | Щороку |
Рання діагностика ФКУ (до 2-місячного віку) та профілактичне лікування (харчування безфенілаланіновою дієтою, наприклад, сумішами: “Берлофен”, “Феніл-Фрі”, “Афенілак”, “Лофеналак”, “Тетрафен”, “Аналог-ХР” “Максамум-ХР”, “Максамаїд-ХР”) попереджують розвиток клінічної картини хвороби. Безфенілаланінові суміші хворим видаються безкоштовно через обласні управління охорони здоров’я. Білок в раціоні дітей становить 20-22% від вікової фізіологічної норми. Недостатня кількість поповнюється за рахунок вище зазначених сумішей. При цьому необхідно враховувати і фенілаланін, який міститься в суміші. Загальна кількість вуглеводнів у раціоні хворої дитини повинна покривати 50-60% загальної калорійності їжі (овочі, фрукти, соки, цукор, крохмаль). Суміш амінокислот та гідролізати білка вводять до раціону поступово. Початкові дози становлять 1/3-1/5 добової кількості препарату. До добової норми ці суміші доводять протягом тижня. Водночас в раціоні зменшують кількість білка природних продуктів. Дітям першого року життя гідролізат білка дається при кожному прийманні їжі, а старшим одного року – двічі на день. При захворюваннях дітей доза гідролізату зменшується або навіть повністю виключається на 1-3 дні. Гідролізат білків, що дається дітям, розводиться кип’яченою водою. У харчуванні дітей старшого віку використовують спеціальні безбілкові продукти, створені на основі пшеничного чи кукурудзяного крохмалю (безбілкові макарони, саго, безбілковий хліб та кондитерські вироби). Кількість жиру дітям збільшують на 10%. Для дієтичної корекції хворих на ФКУ дітей та дорослих використовують таблетки Аврнілу (препарат містить 500 мг амінокислот, збагачений вітамінами та мікроелементами). Препарат використовуються по 1 таблетці за три чи більше прийомів під час їжі.
У 6-8 років хворі уже не потребують спеціальної дієти. Для дітей старше 7 років використовуються для корекції харчування таблетки прекунілу (PreKUnil), які складаються із всіх незамінних амінокислот. Використовуються по 1 таблетці три чи більше разів під час їжі. Дітям старшим 8 років використовуються для дієтичної корекції НеоФЕ (амінокислотна добавка, яка складається із незамінних амінокислот). Одна таблетка містить 500 мг амінокислот.
Діти з фенілкетонурією знаходяться на диспансерному обліку у сімейного лікаря чи педіатра, дитячого невролога.
Гістидинемія – спадкове порушення обміну гістидину, обумовлене відсутністю чи недостатньою активністю гістидази. Тип спадкування аутосомно – рецесивний. У результаті блоку ферменту в організмі здійснюється накопичення гістидину (норма до 2,12 мг/100мл) та його похідних (імідазолпіровиноградної, імідазолоцтової і імідазолмолочної кислот) та зниження концентрації уроканінової, глютамінової та інших кислот. Захворювання зустрічається досить рідко (1:10000 до 1:11000).
Для хворих гістидинемією характерне світле волосся, голубі очі. У клініці на перше місце виступають ураження нервової системи у вигляді зниження інтелекту (ІQ 22-59 од), порушення мови, зміни біоенергетичної активності мозку та судоми. Інколи виявляється гідроцефалія, зміни м’язового тонусу (гіпертонія чи гіпотонія), спастичні парези та паралічі, мозжечкова атаксія, тремор. В ряді випадків захворювання супроводжується анемією, патологією нирок та кісткової системи, аномаліями розвитку. Діти з гістидинемією при нормальному психічному розвитку відрізняються емоційною лабільністю, агресивністю та іншими формами порушення поведінки. Розумова відсталість при гістидинемії не така груба, як при ФКУ. Патологоанатомічна картина при гістидинемії характеризується дилатацією ІІІ шлуночка мозку внаслідок атрофічних змін у ділянці таламуса, наявністю демієлінізації, змінами будови мітохондрій, вираженим астрогліозом.
Діагностика гістидинемії проводиться на основі клініки та визначення рівня гістидину в крові (в 3-10 разів вище від норми) та інших біологічних рідинах (в лікворі). Відмічено, що майже у половини хворих на гістидинемією позитивна проба Феллінга, хоча різко позитивні проби на ФКУ зустрічаються рідко. Підтверджується діагноз визначенням активності гістидази в роговому шарі шкіри чи печінці. Крім того, у цих дітей підвищений рівень в крові та сечі аланіну, низький рівень в крові серотоніну. Використовують для діагностики пробу з пероральним навантаженням гістидином (100 мг хлориду L-гістидину на 1 кг маси дитини в суміші з фруктовим соком з визначенням рівня в сечі до навантаження та через 1, 2, 4, 6, 24 години після навантаження).
Лікування: бідна на білок дієта із низьким рівнем гістидину (16-34 мг/кг).
Гомоцистинурія– в основі захворювання відсутність чи зниження активності ферменту цистатіонінсинтетази, що призводить до порушення обміну метіоніну. Зустрічається рідко (1:50000 до 1:250000).
У хворих в крові підвищується рівень метіоніну та гомоцистину, в сечі з’являється гомоцистин та змішані дисульфіди гомоцистеїну та цистеїну. Щодо піридоксину виділяють дві форми піридоксинчутливу та піридоксинрезистентну гомоцистинурію. Хворі при народженні, як правило, не мають зовнішніх дефектів. Чим старша дитина, тим більш чітко проявляється клініка хвороби.
Для захворювання характерна різноманітність проявів. Найбільш типовим є комплекс симптомів: розумова відсталість, ектопія кристалика, скелетні деформації, тромбоемболії і кардіоваскулярна патологія. Розумова відсталість наявна у половини хворих. Для уражень скелета характерними є: диспропорції у вигляді короткого тулуба, подовження кінцівок, воронкоподібної деформація грудної клітки, вираженого остеопорозу кісток, стигм дисембріогенезу. Основною причиною розумового дефекту при гомоцистинурії є судинні мозкові тромби (в результаті активації фактору Хагемана) і утворення дрібних інфарктів мозку.
Для діагностики гомоцистинурії використовують лабораторні дослідження: визначення цистину і гомоцистину в сечі, визначення метіоніну та цистину в сироватці крові, визначення активності цистатіонінсинтетази в печінці.
Лікування: виключення продуктів, які багаті на метіонін (допускається вміст метіоніну 29-45 мг/кг), призначення вітаміну В6 у великих дозах (більше 30 мг/кг, добова доза до 500 мг).
Цистинурія- характеризується ураженням нирок з порушенням функції проксимальних відділів ниркових канальців. Проявляється підвищеною екскрецією із сечею цистину, а також лізину, аргініну і орнітину. Часто у хворих відмічається піурія.
Успадкування за аутосомно-рецесивним типом. Характеризується утворенням каменів у нирках. Для діагностики має значення позитивна реакція сечі з ціаніднітропрусидом.
Лікування – це призначення дієти, яка не містить метіонін, призначення лужних мінеральних вод. Для профілактики використовується суміш Олбрайт (цитрат калію, лимонна кислота, вода).
Лейциноз(хвороба запаху сечі кленовим сиропом) зустрічається ізчастотою 1-3 : 100000. При цьому синдромі спостерігається ензиматична блокада в процесі декарбоксилування лейцину, ізолейцину та валіну. При цьому утворюється надлишкова кількість кетоізокапронової і кетоізовалеріанової кислот. Тип спадковості – аутосомно-рецесивний. Захворювання проявляється з перших днів життя. Відомі 4 варіанти хвороби: класичний, інтермітуючий, проміжний, тіамін-залежний.
Найбільш частий симптом – це відмова від груді, підвищена збудливість, різкий крик, неритмічне дихання, нападоподібний ціаноз, судоми, м’язова гіпо- чи гіпертонія. Розвиваються ознаки затримки розвитку, пригнічення ЦНС, можливі коматозні стани. Захворювання має важкий перебіг і приводить до загибелі дитини. Характерною діагностичною ознакою є запах сечі, що нагадує кленовий сироп, пивну закваску, м’ясний суп або карамелезований цукор. Для діагностики використовується проба сечі з хлорним залізом, визначення спектру амінокислот у біологічних рідинах. Діагноз підтверджується виявленням дефекту дегідрогенази кетокислот в лейкоцитах.
Для лікування використовуються суміші амінокислот та білкові гідролізати з обмеженою кількістю амінокислот з розгалуженим ланцюгом. Для лікування коматозних станів використовують замінне переливання крові, перитонеальний діаліз. Тіамін-залежна форма піддається лікуванню вітаміном В1, доза якого складає 10 мг/добу і більше.
Цистиноз– захворювання, пов’язане з порушенням обміну цистину і характеризується важким ураженням внутрішніх органів, головним чином нирок. Частота 1:600000. Успадковується за аутосомно-рецесивним типом. Відкладання цистину в різних органах порушує їх функцію.
Клінічно захворювання проявляється в кінці першого року життя. Відмічається погіршення апетиту, блювотиння, закрепи, полідіпсія, поліурія. Характерні підвищення температури без видимих причин. Блювання та поліурія можуть призвести до зневоднення. Типовим проявом захворювання є прогресуюче відставання в рості. Приєднуються подібні до рахіту зміни в кістках: лобні та тім’яні горби, деформація грудної клітки та кінцівок, реберні ”чотки” та “браслетки”. У хворих відмічається м’язова слабкість та гіпотонія, світлобоязнь та явища кон’юнктивіту.
При лабораторному обстеженні виявляється гіпераміноацидурія, фосфатурія, кальційурія. Важливим у діагностиці є обстеження хворих за допомогою щілинної лампи (наявність цистинових кристалів у рогівці).
Лікування: використання вітаміну Д з урахуванням індивідуальної чутливості, призначення нераболу (0,1 мг/кг на добу чи ретаболілу (1 мг/кг на добу) 1 раз в 2-4 тижні по 2,5-3 міс з 2-місячною перервою. Призначається також лужна дієта, введення цитратних сумішей, диметилцистеїн. Прогноз захворювання несприятливий. Смерть наступає в перші роки життя на фоні вираженої ниркової недостатності.
Алкаптонурія (гомогентизинурія).Для захворювання характерна нестача оксидази гомогентизинової кислоти, яка в нормальних умовах сприяє переходу цієї кислоти в малеїлацетооцтову. Тип спадкування - аутосомно-рецесивний.
Клінічно хвороба проявляється трьома симптомами: потемніння сечі (особливо коли стоїть на повітрі чи з добавленням лугу), пігментація хрящів та сполучної тканини (вушні раковини, склери, шкіра носа, рук, шиї), артропатія. Симптоми ураження суглобів, які визначають важкість захворювання, розвиваються після 30-40 років.
Діагноз підтверджується виявленням гомогентизинової кислоти в сечі.
Лікування симптоматичне. Хворим призначають великі дози вітаміну С.
Тирозиноз– спадкове захворювання, яке характеризується порушенням обміну фенілаланіну та тирозину. При тирозинозі порушується перетворення парагідрооксифенілпіровиноградної кислоти в гомогентизинову. Розвивається тирозинемія і тирозинурія. Із сечею виділяється велика кількість парагідрооксифенілмолочної, парагідрооксіфенілпіровиноградної, парагидрооксифенілмолочної кислот. Тип успадкування – аутосомно-рецесивний.
Основними клінічними ознаками захворювання при важких формах є міастенія, блювання, відставання в фізичному розвитку. В крові підвищений рівень тирозину і дещо знижений інших амінокислот. Можливий розвиток цирозу печінки, множинні ураження канальців нефрону (глутамінфосфат – діабет), що супроводжується остеопорозом і остеомаляцією. У деяких хворих спостерігається гіпокаліємія, ацидоз, зниження фосфатів крові. При цьому наявна гіперфосфатурія, глюкозурія, аміноацидурія. В деяких дітей наявна розумова відсталість різного ступеня. У деяких хворих відмічається тромбоцитопенія.
Лікування: зменшення прийому з їжею фенілаланіну та триптофану. Прогноз при дієтотерапії сприятливий.
Читайте також:
- II. Організація і проведення спортивних походів
- II. Організація перевезень
- II. Організація перевезень
- Rete-алгоритм
- А. Організація Острозького колегіуму – Академії
- Адміністративно-територіальна організація
- Алгоритм
- Алгоритм
- Алгоритм 1.
- Алгоритм RLE
- Алгоритм безпосередньої заміни
- Алгоритм Берлекемпа-Мессі
| <== попередня сторінка | | | наступна сторінка ==> |
| Фенілкетонурія | | | Загальна |
|
Не знайшли потрібну інформацію? Скористайтесь пошуком google: |
© studopedia.com.ua При використанні або копіюванні матеріалів пряме посилання на сайт обов'язкове. |