РЕЗОЛЮЦІЯ: Громадського обговорення навчальної програми статевого виховання
ЧОМУ ФОНД ОЛЕНИ ПІНЧУК І МОЗ УКРАЇНИ ПРОПАГУЮТЬ "СЕКСУАЛЬНІ УРОКИ"
ЕКЗИСТЕНЦІЙНО-ПСИХОЛОГІЧНІ ОСНОВИ ПОРУШЕННЯ СТАТЕВОЇ ІДЕНТИЧНОСТІ ПІДЛІТКІВ
Батьківський, громадянський рух в Україні закликає МОН зупинити тотальну сексуалізацію дітей і підлітків
Відкрите звернення Міністру освіти й науки України - Гриневич Лілії Михайлівні
Представництво українського жіноцтва в ООН: низький рівень культури спілкування в соціальних мережах
Гендерна антидискримінаційна експертиза може зробити нас моральними рабами
ЛІВИЙ МАРКСИЗМ У НОВИХ ПІДРУЧНИКАХ ДЛЯ ШКОЛЯРІВ
ВІДКРИТА ЗАЯВА на підтримку позиції Ганни Турчинової та права кожної людини на свободу думки, світогляду та вираження поглядів
- Гідрологія і Гідрометрія
- Господарське право
- Економіка будівництва
- Економіка природокористування
- Економічна теорія
- Земельне право
- Історія України
- Кримінально виконавче право
- Медична радіологія
- Методи аналізу
- Міжнародне приватне право
- Міжнародний маркетинг
- Основи екології
- Предмет Політологія
- Соціальне страхування
- Технічні засоби організації дорожнього руху
- Товарознавство продовольчих товарів
Тлумачний словник
Авто
Автоматизація
Архітектура
Астрономія
Аудит
Біологія
Будівництво
Бухгалтерія
Винахідництво
Виробництво
Військова справа
Генетика
Географія
Геологія
Господарство
Держава
Дім
Екологія
Економетрика
Економіка
Електроніка
Журналістика та ЗМІ
Зв'язок
Іноземні мови
Інформатика
Історія
Комп'ютери
Креслення
Кулінарія
Культура
Лексикологія
Література
Логіка
Маркетинг
Математика
Машинобудування
Медицина
Менеджмент
Метали і Зварювання
Механіка
Мистецтво
Музика
Населення
Освіта
Охорона безпеки життя
Охорона Праці
Педагогіка
Політика
Право
Програмування
Промисловість
Психологія
Радіо
Регилия
Соціологія
Спорт
Стандартизація
Технології
Торгівля
Туризм
Фізика
Фізіологія
Філософія
Фінанси
Хімія
Юриспунденкция
Приклади використання деяких методів аналізу на практиці
При виборі методу, який придатний для аналізу, аналітики зштовхуються з великим набором методів, в яких достатньо важко орієнтуватися. Вибір того чи іншого методу залежить від багатьох факторів: рівня вмісту аналізованого компонента у зразку, вимог точності та чутливості, числа зразків, природи інших речовин присутніх у зразку і, нарешті, вимог до апаратурного оформлення методу.
Визначення пестицидів в продуктах харчування методами
газо-рідинної хроматографії (ГРХ)
В аналізі отрутохімікатів велике розповсюдження одержала ГРХ. Цим методом можуть бути розділені майже всі пестициди, а використання високочутливих та вибіркових детекторів робить можливим визначення навіть мільйонних часток препарату.
Отрутохімікати екстрагують з наважки середньої проби продукту органічними розчинниками (бензен, гексан, хлороформ, етери та ін.) або їх сумішами, інколи водою, так як більшість пестицидів погано у ній розчиняється. При екстракції майже завжди поряд з пестицидами екстрагуються пігменти, жири та інші речовини, що заважають точності визначення, тому екстракт очищують від супутніх речовин методами колоночної та тонкошарової хроматографії, перерозподілом у іншому розчиннику, змішуванням з зневодненою сульфатною кислотою та іншими способами.
Визначення хлорорганічних отрутохімікатів в овочах, фруктах, молоці та продуктах його переробки
Метод ґрунтується на ексракції препаратів (альдріна, гексахлорана, гептахлора, ДДТ, ДДД, ДДЕ та ін.) органічними розчинниками, очистки екстрактів шляхом розподільної хроматографії між двома рідинами, що не змішуються, з наступною очисткою на колонці з алюміній оксидом та розділенні на хроматографі з детектором по захопленню електронів.
Екстракція препаратів та очистка екстрактів. Здрібнені яблука масою 25 г екстрагують діетиловим етером об’ємом 100-150 см3 при струшуванні на протязі 15 хв. Екстракт упарюють досуха, додають попередньо охолоджену до 5-0° С суміш ацетону з водою (4:1), і розчин швидко фільтрують. До фільтрату додають гексан об’ємом 24 см3, водний шар відокремлють, шар гексану сушать над безводним натрій сульфатом, випаровують до об’єму 1-2 см3 та вносять у колонку з алюміній оксидом безводним або ІІ ступеню активності (висота шару сорбенту 7-8 см, на який насипаний шар 3-4 см безводного натрій сульфату). Отрутохімікати елююють з колонки сумішшю етер:н-гексан (10:90) об’ємом 100 см3, зі швидкістю 3-4 краплі у секунду. Елюат упарюють до невеликого об’єму і кількісно переносять у мірну пробірку. Об’єм розчину не повинний перевищувати 1-1,5 см3. Для ГХ-аналізу використовують цей розчин об’ємом 2-4 мксм3.
До молока об’ємом 40 см3 додають ацетон об’ємом 80 см3, н-гексан об’ємом 80 см3 та струшують у конічній колбі на протязі 3 хв. Суміш переносять у стакан для ценрифугування, обмиваючи колбу гексаном об’ємом 10 см3 та водою об’ємом 5 см3, і центрифугують при швидкості 2500 об/хв на протязі 3 хв. Шар гексану відокремлюють, висушують безводним натрій сульфатом масою 8-10 г, декантують та випарюють до об’єму 5-10 см3. Цей розчин кількісно переносять у ділильну лійку, додають диметилформамід або ацетонітрил об’ємом 15-30 см3, насичений гексаном (у співвідношенні 1:2), і струшують на протязі 1 хв. Після цього дають розділитися шарам і переносять шар диметилформаміду або ацетонітрилу у другу ділильну лійку, де знаходиться насичений розчин натрій сульфату об’ємом 350-400 см3 і гексан об’ємом 50 см3 для розчину в диметилформаміді або насичений розчин натрій хлориду об’ємом 350- 400 см3 і гексан об’ємом 50 см3 для розчину в ацетонітрилі. Лійку перевертають 3-4 рази і після розділення шарів шар гексану відокремлюють, а водний шар екстрагують ще раз гексаном об’ємом 50 см3. Гексанові фракції об’єднують, промивають дистильованою водою об’ємом 100 см3, випарюють до об’єму 2-3 см3. Цей розчин очищують на колонці з алюміній оксидом і далі здійснюють операції, які описані для проби яблук.
Хроматографія. Умови розділення наступні: колонка (стальна або скляна) довжиною від 1,5 до 2 м та діаметром від 3 до 6 мм наповнена 10 % SE-301 (або SE-30, або DC-200, або DC-11) на хромосорбі W зернінням 60- 80 меш (обробленому хлоридною кислотою); температура колонки 200° С, детектора 200° С, випарної камери 220° С, швидкість азоту 120 см3/хв; напруга та чутливість встановлюються таким чином, щоб сила струму у комірці детектора була 1,6·10-10 А.
Для кількісного аналізу певний об’єм (2-5 мксм3) стандартного розчину, що містить отрутохімікат масою 0,01-0,025 мкг, розділяють у тих ж умовах, що й пробу. Розрахунок вмісту отрутохімікату у продукті проводять за формулою:
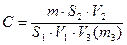 мг/дм3 або мг/кг, де
мг/дм3 або мг/кг, де
m – маса ДДТ у стандартному розчині, мг;
S1 – площа піка стандартного розчину ДДТ, мм2;
S2 – площа піка проби, мм2;
V1 – об’єм екстракту проби, введений у хроматограф, см3;
V2 – загальний об’єм екстракту після випаровування, см3;
V3 (m3) – об’єм або маса наважки аналізованої проби, дм3 або кг.
Визначення залишкових кількостей пестицидів
Метод тонко-шарової хроматографії (ТШХ) для визначення залишкових кількостей пестицидів у харчових продуктах є перспективним з точки зору експресності та вибірковості. Його широко використовують для контролю за залишковим вмістом пестицидів в плодоовочевій сировині і консервній продукції.
Визначення хлорофосу в овочах, фруктах, зерні, молоці, м’ясі
Метод ґрунтується на екстракції отрутохімікату з досліджуваної проби водою з наступним витяганням його з водного розчину хлороформом, очистці екстракту та визначенні в тонкому шарі силікагелю.
Екстракція препарату. Подрібнену пробу (овочі, фрукти, зерно, м’ясо) масою 25 г три рази екстрагують водою об’ємами по 70 см3 та два рази об’ємами по 50 см3 на апараті для струшування на протязі 15 хв. Водні екстракти об’єднують, додають натрій хлорид масою 1-1,5 г (щоб запобігти утворення емульсії при подальшій екстракції) і хлорофос три рази екстрагують хлороформом, який насичений водою, об’ємами по 50 см3. Хлороформні екстракти об’єднують, зливають через шар безводного натрій сульфату та сушать на протязі 10-15 хв.
У випадку аналізу м’яса при струшуванні у ділильній лійці у нижньому хлороформному шарі утворюється стійка емульсія, тому екстракт зливають разом з емульсією. Для її руйнування емульсії до екстракту поступово додають при безперервному перемішуванні безводний натрій сульфат. Пробі дають постояти 10-15 хв, а потім зливають хлороформ через шар безводного натрій сульфату у суху колбу. Колбу з натрій сульфатом, що залишився, промивають чотири рази хлороформом об’ємами по 15 см3 та приєднують до екстракту.
При визначенні хлорофосу у молоці його попередньо знежирюють центрифугуванням на протязі 10 хв при 6000 об/хв та фільтрують через вату. Потім до об’єму 25 см3 додають для звурджування фосфорно-молібденову кислоту масою 1,1 г та залишають на 30 хв, періодично струшуючи. Після звурджування молоко переносять у пробірку для ценрифугування, обмивають колбу дистильованою водою 2-3 рази (об’ємом не більше 20 см3) та центрифугують 10 хв при 6000 об/хв. Верхній шар зливають через вату у ділильну лійку, а до залишку у пробірці для ценрифугування додають дистильовану воду об’ємом 25 см3, розбивають осад паличкою, ретельно перемішують та центрифугують вторинно за тих же умов. Потім об’єднують водні екстракти та додають натрій хлорид масою 1 г. З одержаного розчину препарат три рази витягають хлороформом, що насичений водою, порціями по 40 см3. Хлороформні екстракти зливають через шар безводного натрій сульфату.
Відгонка розчинника. З зневодненого хлороформного екстракту відганяють розчинник досуха (під вакуумом при температурі не вище 40° С) та розчиняють сухий залишок у точно виміряному об’ємі (1 см3) хлороформу або діетилового етеру.
Хроматографія у тонкому шарі силікагелю. На пластинку з сорбентом наносять вказаний розчин об’ємом 0,1 см3, а якщо хлорофосу у пробі міститься мало, то весь розчин. Як сорбент використовують силікагель КСК-3 або ШСК з розміром частинок 40-60 мкм, закріплювач – крохмаль.
На цю ж пластинку по обидві боки від плями проби наносяться “свідки” – стандартні розчини різних концентрацій чистого отрутохімікату. Для додаткової очистки пластинки з нанесеними пробами та “свідками” попередньо занурюють у чистий бензен. Після того як фронт розчинника підніметься на 10 см, пластинку виймають з камери та залишають на декілька хвилин для випаровування розчинника на повітрі. Потім пластинку знову приміщують у камеру, що заповнена рухливим розчинником (гексан з ацетоном у співвідношенні 1:1), та проводять хроматографування, як вказано вище. Висушену пластинку проявляють водним розчином резорцину [ω(резорцину) = 2 %] з водним розчином натрій карбонату [ω(Na2CO3) = = 10 %], які змішують перед обприскуванням у співвідношенні 2:3, з наступним нагріванням при 100° С на протязі 7-10 хв. Хлорофос проявляється у вигляді оранжевої плями.
Кількісне визначення проводять шляхом порівняння розміру та інтенсивності забарвлення плям досліджуваної проби і стандартних розчинів “свідків”. Порівняння проводять або візуально, або шляхом вимірювання їх площі.
Визначення поліциклічних ароматичних вуглеводнів (ПАВ) в продуктах харчування
ПАВ присутні у продуктах рослинного походження та продуктах копчення. В овочах та злакових культурах знайдено 13 ПАВ, які попадають у рослини головним чином з ґрунту. Джерелами попадання ПАВ у харчові продукти слугують також вода, повітря, коптильний дим. У міському повітрі присутні до 15 ПАВ, у тому числі 3,4-бензопирен – речовина сильної канцерогенної дії. Бензопирен (БП) та інші ПАВ знайдені в оліях: оливковій, кукурудзяній, соняшниковій, бавовняній та ін. БП дуже добре розчинний в оліях і концентрується в них в процесі виробництва з плодів та насіння олійних культур.
Найбільша кількість БП знайдена у ліпідах, які містяться у плодовій оболонці насіння олійних культур. Вміст БП у рибі гарячого коптіння, а також коптильних та напівкоптильних ковбасах міститься від 1 мкг до декількох десятків мкг на 1 кг продукту. Високій вміст БП знайдено у тютюновому димі.
Визначення БП та інших канцерогенних ПАВ проводиться за тонкою структурою спектра флуоресценції при низький температурі.
Спектрально-флуоросцентний метод визначення БП включає декілька стадій: екстракції з наважки продукту фракції, яка містить ПАВ; очистку одержаної фракції від домішок і хроматографічне розділення ПАВ; якісне визначення БП та інших ПАВ за спектрами люмінесценції при температурі рідкого азоту; кількісне визначення БП за допомогою одної з модифікацій спектрально-флуоросцентного способу.
Методика визначення БП у коптильних продуктах та рослинних оліях
До фаршу коптильного продукту масою 1 кг приливають етиловий спирт об’ємом 1 дм3, додають калій гідроксид масою 150-250 г (в залежності від вмісту жирів у продукті) і кип’ятять 1,5-2 год для омилення ліпідів. Потім приливають 3-5-кратну кількість дистильованої води та екстрагують неомилені речовини діетиловим етером. Перша порція етеру повинна бути у 4-5 разів більшою за об’ємом розчину, який обробляють. Наступні три-чотири порції етеру повинні бути у 3 рази більше попередньої.
При дослідженні рослинних олій продукт масою 50 г омилюють протягом години на киплячій водяній бані у присутності спиртового розчину калій гідроксиду [ω(KOH) = 25 %] об’ємом 100 см3, потім розчин розводять 10-кратною кількістю дистильованої води та відокремлюють речовини, що не омилилися, багаторазовою екстракцією діетиловим етером.
Етерний екстракт декілька разів промивають дистильованою водою, потім сушать над безводним натрій сульфатом. Етер відганяють, залишок розчиняють у бензені та пропускають через колонку довжиною 120-140 мм, яка заповнена алюміній оксидом. Адсорбовані в колонці ПАВ, відокремлені від інших неомилених речовин, елююють бензеном до того часу, поки перестане виділятися фракція з синьою флуоресценцією. Бензен відганяють з елюату, а залишок фракціонують колоночною або тонкошаровою хроматографією.
Виділену суміш ПАВ, яка містить деякі домішки, розчиняють у петролейному етері об’ємом 10-15 см3 і наносять у заповнену алюміній оксидом колонку діаметром 10-14 мм та висотою 120-140 мм. Єлююють фракції ПАВ, що флуоресціюють, спочатку петролейним етером, потім з додаванням бензену. БП міститься у ІІІ, ІV або V фракціях. Для більш чіткого відокремлення БП можна повторити фракціонування колоночним методом або у тонкому шарі алюміній оксиду. При використання другого методу розчинником є суміш хлороформ : петролейний етер (1:2).
Якісне визначення БП проводять спектральним методом з використанням ефекту Е. В. Шпольського. При температурі -196°С одержують спектри люмінесценції окремих фракцій ПАВ, розчинених у нормальних парафінових вуглеводнях. Для якісного визначення БП використовують суміш, що складається з бензенового екстракту об’ємом 1 см3 та н-октану об’ємом 2 см3. Пробірку з сумішшю вміщують у сосуд Дьюара з рідким азотом. Збуджують люмінесценцію за допомогою ртутно-кварцевої лампи, пропускаючи УФ-випромінювання через фільтр. Для запису спектра звичайно використовують спектрограф ІСП-51 з камерою f=270 мм. У спектрі замороженого н-октанового розчину БП є характерні квазілінії 4030 та 4082 Å.
Кількісне визначення БП флуоресцентно-спектральним методом може бути виконано з використанням будь-якої з модифікацій: за допомогою добавок і встановленням приладу по фону, що створюється домішками, які люмінесціюють і які містяться у досліджуваному екстракті, або за допомогою внутрішнього стандарту.
Гравіметричний метод визначення сульфатів у стічній воді
Воду, що аналізують спочатку фільтрують, потім відбирають аліквоту об’ємом 25-200 см3, в залежності від вмісту сульфатів, про який можна судити за попередньою пробою, переносять у стакан, підкислюють хлоридною кислотою за метиловим оранжевим та випаровують (або розводять) до об’єму 50 см3.
Якщо при упарюванні утворився осад, його відфільтровують через маленький фільтр та промивають гарячою дистильованою водою, яка підкислена хлоридною кислотою. Фільтрат та промивні води знов упарюють до об’єму 50 см3, нагрівають до кипіння та приливають по краплях гарячий розчин барій хлориду [ω(BaCl2) = 5 %] до повного осадження сульфатів. Рідину з осадом, що утворився, залишають стояти на водяній або пісочній бані на протязі 2 год і потім на холоду – ніч. На наступний день осад відфільтровують через щільний фільтр (синя стрічка), промивають гарячою водою до зникнення у промивній воді хлоридів (проба з розчином арґентум нітрату, який підкислений нітратною кислотою), сушать, прожарюють 30 хв та зважують у вигляді барій сульфату та роблять необхідні розрахунки:


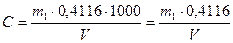 мг/дм3, де
мг/дм3, де
m1 – маса BaSO4,мг;
V – об’єм проби, що взятий для аналізу, см3;
0,4116 – коефіцієнт перерахунку BaSO4 на SO42-.
Визначення вмісту йонів феруму в стічних водах
Ферум у водах може бути в багатьох формах: в істинно розчинному стані та у вигляді колоїдного розчину внаслідок пептизації ферум гідроксиду органічними речовинами; у вигляді комплексних сполук з неорганічними та органічними лігандами, в різних суспензованих у воді твердих частинках. Ферум може бути у тривалентному та двовалентному стані. Необхідно також враховувати те, що у воді, яка містить кисень, ферум (ІІ) легко переходить у ферум (ІІІ) та осаджується у вигляді гідроксиду. Таким чином точні результати можуть бути одержані при визначенні сумарного вмісту феруму у всіх його формах.
Фотометричний метод з натрій сульфосаліцилатом
Метод ґрунтується на тому, що сульфосаліцилова кислота або її натрієва сіль утворюють з солями феруму забарвлені комплексні сполуки, причому, у слабокислому середовищі сульфосаліцилова кислота реагує тільки з солями феруму (ІІІ) (червоне забарвлення), а в слаболужному середовищі – з солями феруму (ІІІ) та (ІІ) (жовте забарвлення).
Пробу об’ємом 100 см3 (чи меншого об’єму, але доведеного до об’єму 100 см3 дистильованою водою), що містить йони феруму масою 0,01-1,0 мг, вміщують у хімічний стакан, додають концентрованої нітратної кислоти об’ємом 0,5 см3 і упарюють на електроплитці приблизно вдвічі. Концентрований розчин розводять водою і відфільтровують, збираючи фільтрат у мірну колбу об’ємом 100 см3. Фільтр промивають, а фільтрат у колбі доводять до об’єму близько 80 см3. До обробленої таким чином проби приливають піпетками розчин амоній хлориду об’ємом 2 см3, розчин сульфосаліцилової кислоти об’ємом 2 см3, розчин амоній гідроксиду об’ємом 2 см3, перемішують суміш і дистильованою водою доводять об’єм у мірній колбі до 100 см3, знову перемішують. Через 5 хв вимірюють оптичну густину за відношенням до холостої проби (світлофільтр λ=420-430 нм). За калібрувальним графіком знаходять вміст йонів феруму у пробі та розраховують за формулою:
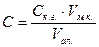 мг/дм3, де
мг/дм3, де
Cк.г. – вміст йонів феруму, що знайдений за калібрувальним графіком, мг/см3;
Vм.к. – об’єм мірної колби, 100 см3;
Vал. – об’єм взятої проби для аналізу, см3.
Побудова калібрувальної кривої
У мірні колби об’ємом 100 см3 піпетками точно відмірюють об’єми робочого стандартного розчину 0; 1,0; 2,0; 10,0; 20,0; 40,0; 60,0; 80,0; 100,0 см3 [стандартний розчин C(Fe3+) = 0,1 мг/см3, що розведений у 10 разів]. У еталонах визначають йони феруму методом, що описаний вище. Оптичну густину визначають за вибраним світлофільтром по відношенню до холостої проби (холостою пробою є перший розчин еталонної серії, куди стандартний розчин феруму не вводили). За одержаними даними будують графік в координатах “оптична густина – масова концентрація феруму”.
Вміст йонів феруму у кожному стандартному розчині визначають за формулою:
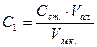 мг/дм3, де
мг/дм3, де
Cк.г. – вміст йонів феруму у робочому стандартному розчині, мг/см3;
Vм.к. – об’єм мірної колби, 100 см3;
Vал. – об’єм взятої проби для аналізу, см3.
Визначення хлорид-іонів в стічних водах титриметричними методами
Меркуриметричний метод
Хлорид-іони утворюють з йонами Меркурію малодисоційований меркурій хлорид HgCl2. Як індикатор використовують дифенілкарбазон, який утворює наприкінці титрування з надлишком йонів Hg2+ комплексну сполуку забарвлену у фіолетовий колір. Титрування проводять у середовищі нітратної кислоти при рН = 2,5±0,1.
Відбирають профільтровану пробу об’ємом 100 см3 (або менший її об’єм, розводялячи дистильованою водою до об’єму 100 см3), яка містить в цьому об’ємі хлорид-іони масою 0,5-10 мг, і додають розчин індикатора об’ємом 0,3 см3. Потім для встановлення рН = 2,5 додають по краплях нітратну кислоту [С(1/1 HNO3) = 0,2 моль/дм3] до того часу, поки колір розчину не перейде з синього у жовтий, і ще кислоти об’ємом 1,0 см3. При аналізі сильнокислих проб розчин при додаванні індикатора забарвлюється у жовтий колір, тоді додають по краплях розчин натрій гідроксиду [С1/1 NaOH) = 0,1 моль/дм3] до переходу забарвлення у синє-зелене і після цього вводять нітратну кислоту [С1/1 HNO3) = 0,2 моль/дм3] об’ємом 1,0 см3. Встановивши необхідне значення рН, титрують розчином меркурій (ІІ) нітратом до переходу жовтого забарвлення у фіолетове.
Якщо треба дуже точне визначення вмісту хлорид-іонів, проводять холостий дослід з дистильованою водою об’ємом 100 см3.
Вміст хлорид-іонів розраховують за формулою:
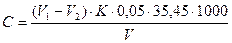 (мг/дм3), де
(мг/дм3), де
V1 – об’єм розчину меркурій нітрату, що витрачений на титрування проби, см3;
V2 – об’єм розчину меркурій нітрату, що витрачений на титрування холостого досліду, см3;
К – поправочний коефіцієнт для приведення молярної концентрації еквівалента розчину меркурій нітрату до точно 0,05 моль/дм3;
0,05 – молярна концентрація еквіваленту розчину меркурій нітрату С(1/2 Hg(NO3)2) = 0,05 моль/дм3;
35,45 – молярна маса еквівалента хлорид-іону, г/моль;
V – об’єм проби, см3.
Визначення нафтопродуктів у стічних водах методом
ІЧ-спектрометрії
При використання цього методу немає необхідності відгонки розчинника після екстракції нафтопродуктів розчинником зі стічної води. Як екстрагент використовують тетрахлорометан (CCl4).
Пробу стічної води 1-5 дм3, в залежності від вмісту нафтопродуктів, підкислюють до рН = 2, додають натрій хлорид масою по 2 г на кожний літр рідини та проводять екстракцію CCl4 у ділильній лійки, додають розчинник окремими порціями, перемішуючи перевертанням лійці на протязі 10 хв після додавання кожної порції, витримують до розшарування 10 хв та збирають екстракт у колбу з скляною пробкою. Всього на екстрагування витрачається екстрагент об’ємом 60 см3. Екстракт висушують прожареним натрій сульфатом, відбирають аліквоту об’ємом 50 см3 і пропускають її через колонку з алюміній оксидом. Збирають елюат у мірну колбу ємністю 100 см3. Пропускають через колонку ще чистий CCl4 об’ємом 45 см3 і доводять цим же розчинником об’єм розчину у колбі до мітки.
Знімають ІЧ-спектр одержаного розчину, використовуючи кювету товщиною 50 мм. Вимірюють оптичну густину при хвильовому числі 2926 см-1.
Вміст нафтопродуктів знаходять за формулою:
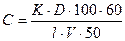 (мг/дм3), де
(мг/дм3), де
К – коефіцієнт, що дорівнює 0,437 або 0,542;
D – оптична густина;
100 – об’єм CCl4 після розведення, см3;
60 – об’єм CCl4, що взятий для екстракції, см3;
l – товщина шару в кюветі, см;
V – об’єм стічної води, що узятий для аналізу, см3;
50 – об’єм аліквотної частини, см3.
Визначення концентрації хлору у повітрі
У процесі аналізу повітря та інших газових сумішей велике значення мають способи відбору газових проб для дослідження. Найбільш розповсюдженим способом відбору проб повітря на аналіз є аспіраційний. Сутність цього методу полягає у просмоктуванні досліджуваного повітря через поглинальні розчини за допомогою приладу (аспіратора) з одночасним вимірюванням об’єму повітря, пропущеного через поглинач. Об’єм води, що витікає з аспіратора, і, відповідно об’єм повітря, що проходить через поглинач, вимірюють мірним циліндром. Як поглиначі використовують розчини таких речовин, які поглинають домішки у повітрі або іншій газовій суміші. При визначенні хлору у повітрі йодометричним методом використовують водний розчин калій йодиду.
У сосуд з повітрям та хлором невеликими порціями (об’ємом по 20- 25 см3) через крапельну або звичайну лійку і при обережному перемішуванні круговими рухами вводять розчин калій йодиду [ω(KI) = 1,5-2%] об’ємом 200-250 см3. Такої кількості калій йодиду достатньо для взаємодії з хлором, який знаходиться у колбі.
З розчину, що утворився, відбирають піпеткою об’єми 20, 25 або 50 см3, переносять у колбу для титрування і титрують розчином натрій тіосульфату [С(1/2 Na2S2O3) = 0,1 моль/дм3] до переходу забарвлення з темно-жовтого до слабо-жовтого. Після цього додають 4-5 крапель крохмального розчину і знову титрують розчин до зникнення синього забарвлення.
Об’ємну частку хлору у повітрі знаходять за формулою:
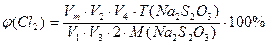 , де
, де
V1 – об’єм сосуду з повітрям та хлором, см3;
V2 – об’єм розчину калій йодиду, що введений у сосуд, см3;
V3 – об’єм аліквоти розчину йоду, що утворився, см3;
V4 – середній об’єм розчину натрій тіосульфату, що витрачений на титрування розчину йоду, см3;
Vm– молярний об’єм, см3;
Т(Na2S2O3) – титр розчину натрій тосульфату, мг/см3;
М(Na2S2O3) – молярна маса натрій тосульфату, г/моль.
Визначення сульфур (ΙV) оксиду у повітрі
Сульфур (ІV) оксид (SO2) поглинають з атмосферного повітря динатрійтетрахлоромеркуратом (ІІ) (Na2HgCl4) розчином, який зв’язує SO2 у формі натрій дихлоросульфітмеркурата (ІІ), запобігаючи повторного потрапляння SO2 у потік газу. Аналіз закінчується додаванням кислого п-розаніліна та формальдегіду до комплексного меркурат-іона, в результаті чого утворюється червона п-розанілінова метилсульфокислота, концентрація якої визначається спектрофотометрично.
Встановлюють пробовідбірну систему, яка складається з повітряного насосу, витратоміру, термометра, малогабаритного імпінджера, що містить 10 см3 поглинаючого розчину. Тиск потрібно вимірювати витратоміром. Повітря прокачується через систему зі швидкістю 0,2-2,5 дм3/хв, поки не буде поглинена необхідна кількість SO2.
Після відбору проби фільтрують розчин, якщо є будь-який осад і доводять об’єм до 10 см3. Додають розчин кислого п-розаніліна [ω(п-розаніліна) = 0,04%] об’ємом 1,0 см3 та розчин формальдегіду [ω(HCHO) = 0,02%] об’ємом 1,0 см3, перемішують і залишають на 20 хв.
Повинен бути підготовлений контрольний розчин аналогічною обробкою Na2HgCl4 [С(Na2HgCl4) = 0,1 моль/дм3] об’ємом 10 см3, виключаючи експозицію у струмені повітря. Величина поглинання розчину визначається при довжині хвилі 560 нм з використанням контрольного розчину для порівняння.
Визначення нітрат-іонів у ґрунті
Нітратна форма Нітрогену у ґрунті дуже рухома. Вона не зв’язується хімічно, не адсорбується ґрунтом і може бути зв’язана тільки біологічно. Вміст нітратів у ґрунті протягом вегетаційного періоду змінюється. Нітратний Нітроген є основним елементом живлення рослин, а тому його визначення за певними фазами має велике значення. Солі нітратної кислоти легкорозчинні, а тому для їх визначення готують водну витяжку.
Визначення нітрат-іонів у ґрунті за допомогою дисульфофенолової кислоти
Наважку ґрунту масою 20 г зважують на технохімічних терезах, потім переносять у плоскодонну колбу об’ємом 250 см3, додають дистильовану воду об’ємом 100 см3 і збовтують протягом 3 хв. Далі фільтрують через складчастий фільтр. Перші порції фільтрату не потрібні. Витяжка має бути прозорою. Коли ґрунти багаті на органічні речовини, витяжка часто буває каламутною. Щоб витяжка завжди була прозорою, у колбу додають перед збовтуванням (на кінчику ножа) алюмокалієвих галунів для осадження колоїдів ґрунту та кращого фільтрування.
Після цього відбирають піпеткою аліквоту об’ємом 25 або 50 см3 фільтрату у фарфорову чашку і випарюють досуха на водяній бані.
До сухого залишку додають дисульфофенолової кислоти об’ємом 1,0 см3, яка розчиняє осад. Скляною паличкою з оплавленим кінцем добре розтирають осад по всій внутрішній поверхні чашки.
Через 10 хв додають воду об’ємом 10-15 см3, перемішують, нейтралізують розчином лугу [ω(NaOH чи KOH) = 10%] до утворення лужної реакції, яка визначається за допомогою лакмусового паперу. Якщо у лужному середовищі є нітрати, розчин забарвлюється у жовтий колір, коли ж нітратів нема, розчин не забарвлюється у жовтий колір.
Якщо рідина пожовтіла, її з чашечки переносять у мірну колбу об’ємом 50 або 100 см3 (в залежності від інтенсивності забарвлення), ополіскують 2-3 рази чашку водою і додають дистильованої води до риски. Рідину у колбі треба перемішати, а потім колориметрувати.
При інтенсивному забарвленні розчин можна розводити в 2 і більше разів, а під час розрахунків це враховувати.
Щоб виготовити стандартний розчин, в окремих фарфорових чашках одночасно з випарюванням досліджуваного розчину випарюють розчин певної концентрації (стандартний розчин) об’ємоми 1; 2; 5; 10; 15; 20; 30 см3 і повторюють ті самі операції, що й з зразками, що досліджуються.
Потім вимірюють оптичну густину приготовлених стандартних розчинів (при синьому світлофільтрі) і будують калібрувальний графік.
Визначивши величину оптичної густини досліджуваного розчину, за калібрувальним графіком знаходять відповідну концентрацію, а потім за формулою обчислюють вміст нітратів:
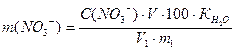 мг, де
мг, де
m(NO3-) – вміст нітрат-іонів, мг на 100 г абсолютно сухого ґрунту;
С(NO3-) – вміст вміст нітрат-іонів, знайдений за калібрувальним графіком, мг на 100 см3;
V – загальний об’єм фільтрату, см3;
V1 – об’єм витяжки, взятої для випаровування, см3;
m1 – наважка ґрунту, яку взято для приготування фільтрату, г;
100 – коефіцієнт перерахунку на 100 г ґрунту;
КH2O – коефіцієнт перерахунку на сухий ґрунт.
Виявлення етилового спирту в напоях і розчинах методом газо-рідинної хроматографії
Для виявлення етилового спирту в розчинах, напоях та інших рідинах методом газо-рідинної хроматографії як еталонну речовину використовують етиловий спирт з масовою часткою етанолу 95 %. Перед введенням у дозатор хроматографа цей спирт переводять у більш летку, ніж етиловий спирт (Ткип.= 78º С), сполуку етилнітрат (Ткип.= 17º С). Для цього до етилового спирту додають натрій або калій нітрат (ІІІ) і трихлороцтову кислоту. Етилнітрат, що утворився, перебуває у вигляді газу над рідиною, його вводять у газовий хроматограф і здійснюють хроматографування.
Умови хроматогрофії: хроматограф, оснащений катарометром; металева колонка довжиною 100 см, діаметром 0,6 см; твердий носій: сферохром, хезасорб чи інші носії; нерухома рідка фаза: поліетиленгліколь (М = 1000-1500 г/моль), нанесений на твердий носій у кількості 12 %; температура термостатів колонки та детектора 75º С, температура детектора – кімнатна; газ-носій – технічний азот, який пропускають через хроматографічну колонку з швидкістю 50-60 см3/хв; струм детектора: 60- 100 мА; швидкість руху діаграмної стрічки: 720 мм/год.
У флакон з-під пеніциліну вносять розчин трихлороцтової кислоти [ω(CCl3COOH) = 50 %] об’ємом 0,5 см3 та водний розчин еталонної речовини (етиловий спирт з масовою часткою 95 %, розведений таким чином, щоб масова частка розчину складала 3-4 ‰) об’ємом 0,5 см3. Флакон закривають гумовою пробкою і закріплюють спеціальним пристроєм (фіксатором). Потім за допомогою шприца через гумову пробку у флакон вводять розчину натрій нітрату (ІІІ) [ω(NaNO2) = 30 %] об’ємом 0,25 см3. Вміст флакона протягом 1 хв ретельно перемішують і за допомогою іншого сухого шприца набирають газову фазу, що знаходиться над рідиною, об’ємом 3 см3. Її вводять у дозатор хроматографа і здійснюють хроматографування. При цьому записують час отримування етилнітрату.
Після закінчення хроматографування еталонної речовини проводять точно такий самий дослід з розчином, у якому передбачають наявність етилового спирту.
Збіг часу утримання речовини в обох пробах (у пробі з еталонною і досліджуваною речовиною) свідчить про ідентичність цих речовин.
Методика виявлення етилового спирту в крові та сечі аналогічна методиці виявлення його в напоях та розчинах.
Кількісне визначення етилового спирту в крові та сечі методом газорідинної хроматографії
Для кількісного визначення етилового спирту в крові та сечі застосовують метод внутрішнього стандарту як один з методів газо-рідинної хроматографії. Згідно з цим методом, до крові або сечі, в яких визначають кількісний вміст етилового спирту, додають внутрішній стандарт. Як внутрішній стандарт застосовують пропіловий спирт. Етиловий спирт, що міститься в крові або сечі (Ткип.= 78º С), а також пропіловий спирт (Ткип.= 97,5º С), який внесли як внутрішній стандарт, переводять у більш леткі сполуки етилнітрат (Ткип.= 17º С) і пропілнітрат (Ткип.= 46-48º С). Суміш етилнітрату та пропілнітрату вводять у дозатор хроматографу і здійснюють хроматографування. При цьому на хроматограмі виписуються два піки, один з них відповідає етиловому спирту (етилнітрату), другий – пропіловому спирту (пропілнітрату). Потім обчислюють відношення площі або висоти піка етилового спирту (етилнітрату) до площі або висоти піка пропілового спирту (пропілнітрату). Розрахунок кількісного вмісту етилового спирту в крові та сечі здійснюють за калібрувальним графіком.
Побудова калібрувального графіка.Спочатку готують серію стандартних розчинів, які містять по 2, 3, 4, 5 ‰ етилового спирту, і розчин внутрішнього стандарту, в якому міститься 4 ‰ пропілового спирту. У кілька флаконів з-під пеніциліну вносять внутрішній стандарт (пропіловий спирт) об’ємом по 2 см3, потім у кожний флакон додають етиловий спирт різної концентрації (2, 3, 4, 5 ‰) об’ємом по 2 см3. Вміст флаконів ретельно перемішують, з кожного флакона беруть суміш спиртів об’ємом по 1 см3 і переносять в інші флакони з-під пеніциліну. У кожній флакон додають розчин трихлороцтової кислоти [ω(CCl3COOH) = 50 %] об’ємом по 0,5 см3. Флакони закривають гумовими пробками, які закріплюють фіксаторами. Потім за допомогою шприца через гумову пробку у флакони вводять розчин натрій нітрату (ІІІ) [ω(NaNO2) = 30 %] об’ємом по 0,25 см3. Вміст флаконів збовтують протягом 1 хв. Після цього за допомогою іншого шприца з кожного флакона беруть газову фазу, яка знаходиться над рідиною, об’ємом по 3 см3 і вводять її у хроматограф, дотримуючись умов хроматографування, зазначених вище.
На хроматограмах вимірюють висоти або площі піків, обчислюють відношення площ або висот піків етилового спирту до площ або висот піків пропілового спирту для кожної проби. Одержані результати множать на 100 і наносять на вісь ординат калібрувального графіка, на вісь абсцис наносять концентрації етилового спирту (в ‰).
Визначення етилового спирту в крові та сечі. У флакон з-під пеніциліну вносять розчин внутрішнього стандарту (пропілового спирту, концентрація якого складає 4 ‰) об’ємом по 2 см3, додають суміш крові об’ємом 2 см3 або сечі, які досліджують на наявність етилового спирту. Вміст флакона ретельно збовтують, потім рідину (суміш крові або сечі з внутрішнім стандартом) об’ємом 1 см3 переносять в іншій флакон з-під пеніциліну і додають розчин трихлороцтової кислоти [ω(CCl3COOH) = 50 %] об’ємом 0,5 см3. Флакон закривають гумовою пробкою і закріплюють фіксатором. За допомогою шприца через пробку у флакон вносять розчин натрій нітрату (ІІІ) [ω(NaNO2) = 30 %] об’ємом 0,25 см3. Вміст флакона ретельно збовтують протягом 1 хв. Потім за допомогою іншого сухого шприца відбирають з флакона газову фазу об’ємом 3 см3, яку переносять у дозатор хроматографа, і здійснюють хроматографування.
На хроматограмі визначають площі або висоти піків і обчислюють відношення площі або висоти піка етилового спирту до площі або висоти піка внутрішнього стандарту. На основі цього співвідношення, помноженого на 100, за калібрувальним графіком розраховують вміст етилового спирту в крові та сечі розчину натрій нітрату (ІІІ) (в ‰).
При визначенні етилового спирту в крові знайдену за калібрувальним графіком концентрацію цього спирту множать на 0,95, у сечі знайдену концентрацію етилового спирту множать на 1,05.
Читайте також:
- H) інноваційний менеджмент – це сукупність організаційно-економічних методів управління всіма стадіями інноваційного процесу.
- XIII. Використання амортизаційних відрахувань
- А. Розрахунки з використанням дистанційного банкінгу.
- Аверсивную терапію використовують, як правило, при лікуванні алкоголізму, нікотиновій залежності і деяких інших захворювань.
- Автоматизовані системи управлінні охороною праці, обліку, аналізу та дослідження травматизму
- Автоматизовані інформаційні системи для технічного аналізу товарних, фондових та валютних ринків.
- Алгебра та початки аналізу
- Алгоритм однофакторного дисперсійного аналізу за Фішером. Приклад
- Альтернативна вартість та її використання у проектному аналізі
- АЛЬТЕРНАТИВНІ ПІДХОДИ ДО ВИДІЛЕННЯ МЕТОДІВ УПРАВЛІННЯ
- Аналіз використання капіталу.
- Аналіз використання матеріальних ресурсів
| <== попередня сторінка | | | наступна сторінка ==> |
| Фізико-хімічні методи аналізу | | | Література |
|
Не знайшли потрібну інформацію? Скористайтесь пошуком google: |
© studopedia.com.ua При використанні або копіюванні матеріалів пряме посилання на сайт обов'язкове. |