РЕЗОЛЮЦІЯ: Громадського обговорення навчальної програми статевого виховання
ЧОМУ ФОНД ОЛЕНИ ПІНЧУК І МОЗ УКРАЇНИ ПРОПАГУЮТЬ "СЕКСУАЛЬНІ УРОКИ"
ЕКЗИСТЕНЦІЙНО-ПСИХОЛОГІЧНІ ОСНОВИ ПОРУШЕННЯ СТАТЕВОЇ ІДЕНТИЧНОСТІ ПІДЛІТКІВ
Батьківський, громадянський рух в Україні закликає МОН зупинити тотальну сексуалізацію дітей і підлітків
Відкрите звернення Міністру освіти й науки України - Гриневич Лілії Михайлівні
Представництво українського жіноцтва в ООН: низький рівень культури спілкування в соціальних мережах
Гендерна антидискримінаційна експертиза може зробити нас моральними рабами
ЛІВИЙ МАРКСИЗМ У НОВИХ ПІДРУЧНИКАХ ДЛЯ ШКОЛЯРІВ
ВІДКРИТА ЗАЯВА на підтримку позиції Ганни Турчинової та права кожної людини на свободу думки, світогляду та вираження поглядів
- Гідрологія і Гідрометрія
- Господарське право
- Економіка будівництва
- Економіка природокористування
- Економічна теорія
- Земельне право
- Історія України
- Кримінально виконавче право
- Медична радіологія
- Методи аналізу
- Міжнародне приватне право
- Міжнародний маркетинг
- Основи екології
- Предмет Політологія
- Соціальне страхування
- Технічні засоби організації дорожнього руху
- Товарознавство продовольчих товарів
Тлумачний словник
Авто
Автоматизація
Архітектура
Астрономія
Аудит
Біологія
Будівництво
Бухгалтерія
Винахідництво
Виробництво
Військова справа
Генетика
Географія
Геологія
Господарство
Держава
Дім
Екологія
Економетрика
Економіка
Електроніка
Журналістика та ЗМІ
Зв'язок
Іноземні мови
Інформатика
Історія
Комп'ютери
Креслення
Кулінарія
Культура
Лексикологія
Література
Логіка
Маркетинг
Математика
Машинобудування
Медицина
Менеджмент
Метали і Зварювання
Механіка
Мистецтво
Музика
Населення
Освіта
Охорона безпеки життя
Охорона Праці
Педагогіка
Політика
Право
Програмування
Промисловість
Психологія
Радіо
Регилия
Соціологія
Спорт
Стандартизація
Технології
Торгівля
Туризм
Фізика
Фізіологія
Філософія
Фінанси
Хімія
Юриспунденкция
Підгрупи II групи
Деякі властивості елементів та простих речовин побічної
| Назва елемента | Хі-міч- ний сим-вол | Будова зов-нішнього і передостан нього елек- тронних шарів атома | Радіус атома, нм | Енергія йоніза-ції Е → E+ еВ | Густина, г/см3 | Температура плавлення, °С | Темпе-ратура кипіння, °С | Стандартний електродний потенціал процесу E2+→ E, В |
| Цинк | Zn | 3s23p63d 104s2 | 0,139 | 9.36 | 7.13 | 419.5 | -0.763 | |
| Кадмій | Cd | 4s24p64d 105s2 | 0.156 | 8.99 | 8.65 | 321.0 | -0.403 | |
| Меркурій | Hg | 5s25p65d 106s2 | 0.160 | 10.44 | 13.55 | -38.89 | 356.7 | +0.850 |
Наявністю d10-підрівня на передостанньому енергетичному рівні атомів елементів підгрупи Цинку спричинена аномальна зміна радіусів їхніх атомів.
Активність елементів підгрупи Цинку збільшується знизу вгору.
Особлива стійкість псевдоінертногазової 6s2-електронної конфігурації зумовлює дуже високий потенціал йонізації Меркурію, він вищий, ніж у всіх решти d -елементів. Цією особливістю Меркурію і пояснюється його істотна відмінність від Цинку і Кадмію. Так, на відміну від Цинку і Кадмію, існує ряд похідних йона Нg , в яких атоми Меркурію сполучені між собою ковалентними зв'язками Нg—Нg, тобто знову виникає псевдоінертногазова конфігурація 6s2. Йони Нgстійкі у водних розчинах.
, в яких атоми Меркурію сполучені між собою ковалентними зв'язками Нg—Нg, тобто знову виникає псевдоінертногазова конфігурація 6s2. Йони Нgстійкі у водних розчинах.
Вміст Цинку у земній корі становить 2∙10-2%. У вільному стані Zn як активний метал у природі не існує. Трапляється він у вигляді цинкової обманки ZnS, значно рідше — у вигляді мінералу галмею, або смітсоніту, ZnСО,. Цинк входить до складу деяких рослин (у подорожнику його -0,02 %, у фіалці -0,05%). У людському організмі Цинк накопичується у зубах (-0,02 %). Цинк виявлено в складі панцирів деяких тварин, наприклад черепах.
Природний цинк — це суміш п'яти стабільних ізотопів з масовими числами 64, 66—68, 70.
Для добування цинку збагачений концентрат 2п8 випалюють:
2ZnS + 3О2= 2ZnО + 2SО2.
Оксид цинку, що утворився, відновлюють вуглецем:
ZnО + С = Zn + СО.
Якщо цинкові руди містять невеликі кількості цинку, то їх переробляють гідрометалургійним способом. Руду, що містить ZnО, випалюють, а потім обробляють розбавленим розчином Н2SО4:
ZnО + Н2SО4 = ZnSО4 + Н2О.
Добутий розчин ZnSО4 піддають електролізу: на алюмінієвих катодах виділяється цинк, аноди виготовлені із свинцю, в процесі електролізу вони не руйнуються. Завдяки низькій температурі кипіння (906°С) цинк можна очищати перегонкою.
Світовий видобуток цинку досягає кількох мільйонів тонн на рік.
Цинк—голубувато-сріблястий метал, досить м'який, крихкий, кристалізується в гексагональних ґратках, плавиться за температури 419°С. Цинк на повітрі вкривається захисною плівкою (плівка містить і карбонат).
Цинк знаходить широке застосування. Ним вкривають поверхні залізних і стальних виробів, його використовують для виготовлення сплавів (латуні, дюралю), друкарських кліше, гальванічних елементів. Цинк застосовується як відновник під час добування силіцію, бору.
Цинк, згідно з його розміщенням у ряду електрохімічних потенціалів, належить до хімічно активних металів, він легко розчиняється у кислотах і лугах:
Zn + 2НСІ + 4Н2О = [Zn(Н2О)4]СІ2 + Н2↑;
Zn + 2NaОН + 2Н2О =Na2[Zn(ОН)4] + Н2↑.
Вода майже не діє на цинк, хоча він стоїть у ряду електрохімічних потенціалів до водню. Пояснюється це тим, що гідроксид цинку, який утворюється на поверхні цинку під час взаємодії його з водою, практично не розчиняється у воді.
Під час нагрівання цинкового пилу в кисні цинк займається і горить зеленкувато-білим полум'ям з утворенням оксиду ZnО білого кольору. Оксид цинку досить стійкий проти дії води і повітря, тому його використовують як білу фарбу (цинкове білило), а також вводять до складу пудри. Значна кількість ZnО використовується у фармацевтичній промисловості для виготовлення присипок, гігієнічних паст, мазей. ZnО застосовується як каталізатор синтезу органічних речовин.
Оксид цинку у воді не розчиняється, відповідний йому гідроксид Zn(ОН)2 можна добути непрямим способом під дією на розчинні солі Цинку лугів.
Zn(ОН)2, як і ZnО,— амфотерна сполука. Взаємодіючи з розчинами лугів, утворює гідроксоцинкати (наприклад, Na2[Zn(ОН)4]), під час сплавляння з лугами або основними оксидами — цинкати (Ме2ZnO2).
Завдяки утворенню гідроксоцинкатів, а також згідно з положенням у ряду електрохімічних потенціалів, цинк взаємодіє з розчинами лугів із виділенням водню.
Цинк здатний до комплексоутворення. Особливо стійкими є комплексні аміакати цинку. Гідроксид цинку розчиняється в аміаку з утворенням комплексного йона [Zn(NН3)4]2+:
Zn(ОН)2 + 4NH3 = [Zn(NН3)4](ОН)2.
Внаслідок цієї реакції утворюється комплексна основа, яку за ступенем дисоціації можна віднести до лугів.
Цинк виявляє підвищену реакційну здатність до галогенів і сірки.
Галогеніди цинку добувають прямим синтезом, а також під час взаємодії оксиду цинку з галогеноводневими кислотами. Сполуки типу ZnГ2 добре розчиняються у воді за винятком ZnF2. Галогеніди цинку мають сольову природу і як солі слабкої кислоти у водних розчинах сильно гідролізують.
Хлорид цинку на повітрі розпливається, приєднуючи дві молекули води, і набуває властивостей кислоти:
ZnС12 + 2Н2О = Н2[Zn(ОН)2СІ2].
Концентрований розчин хлориду цинку («травлена» хлоридна кислота) використовується для очищення поверхонь металів під час паяння.
Кислота Н2[Zn(ОН)2С12] здатна розчиняти клітковину, тому концентровані розчини 2пС12 використовуються у виробництві рослинного пергаменту.
Під дією аміаку на галогеніди цинку утворюються амінокомплекси:
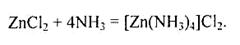 |
Прямим синтезом або під дією сірководню на розчини солей Цинку можна добути білий сульфід цинку ZnS. Він існує у двох кристалічних модифікаціях, використовується як складова частина малярської фарби ліпотон. ZnS у воді не розчиняється.
Завдяки акцепторній здатності йонів Zn2+ Цинк утворює ряд комплексних сполук. Для комплексних сполук Цинку характерне координаційне число 4 і тетраедричне розміщення лігандів. Можуть утворюватись також комплексні сполуки Цинку з координаційним числом 6. Найбільш поширені аміакати та галогеніди цинку.
З аміаком і водою йони Цинку утворюють катіони [Zn(Н2О)4]2+ [Zn(NH3)4]2+та нейтральні [ZnС12∙2Н2О], [ZnС12 ∙NH3] комплекси. Досить міцними є аквакомплекси Цинку. Саме тому з водних розчинів виділяються кристалогідрати розчинних солей Цинку: Zn(NО3)2 ∙ 6Н2О, ZnSО4 ∙ 7Н2О, ZnSО3 ∙7Н2О тощо.
Із водних розчинів галогенідів та псевдогалогенідів цинку можна виділити комплексні солі К2[Zn(СN)4], Сs2[ZnС14].
Слід зазначити, що здатність Цинку до комплексоутворення нижча, ніж його аналогів — Кадмію і Меркурію.
Вміст Кадмію у земній корі становить 1,3∙10-5%. У вільному стані Кадмій не трапляється. Основний мінерал Кадмію — сульфід СdS, частіше Кадмій як домішка входить до складу поліметалічних руд.
Для Кадмію відомо вісім ізотопів з масовими числами 106, 108, 110—114, 116, з яких найпоширенішими є: 112Сd(24,1%) та 114Сd(28,9%). Ядра атомів Кадмію здатні активно поглинати теплові нейтрони.
Кадмій у промисловості добувають з відходів цинкового виробництва. Для цього їх обробляють сульфатною кислотою, а потім витісняють металічний кадмій з його сульфату цинком:
СdSО4 + Zn = ZnSО4 + Сd
Металічний кадмій очищають від цинку, переплавляючи його під шаром розплавленого лугу, який з цинком утворює цинкат. Кадмій високого ступеня чистоти добувають вакуумною дистиляцією. Щорічне світове виробництво кадмію досягає десятків тисяч тонн.
Кадмій — сріблясто-білий метал, м'який, ковкий, в'язкий, кристалізується в гексагональних ґратках, плавиться за температури 321°С. Кадмій стійкий проти дії повітря завдяки наявності оксидної плівки, що утворюється на поверхні металу.
Кадмій використовується для нанесення захисних покриттів на поверхні залізних і стальних виробів. Кадмій, на відміну від цинку, стійкий проти дії лугів. Близько 10 % кадмію йде на виробництво сплавів. Кадмій завдяки великому поперечному перерізу захоплення нейтронів використовується для виготовлення аварійних стержнів для ядерних реакторів.
Кадмій досить активний метал, однак його активність нижча, ніж цинку. У сполуках Кадмій виявляє ступінь окиснення тільки +2.
Кадмій у лугах практично не розчиняється, а в кислотах розчиняється менш енергійно, ніж цинк. У розбавлених розчинах НС1 і Н2SО4 кадмій розчиняється з виділенням водню досить повільно:
Сd + 2НС1 = СdСl2 + Н2↑,
у розбавленому розчині нітратної кислоти — значно краще:
0 +5 +2 +1
4Сd + 10HNO3 = 4Сd(NO3)2 + N2O↑ + 5Н2О.
Під час нагрівання кадмій інтенсивно взаємодіє з активними неметалами, в атмосфері кисню горить з утворенням оксиду Сd, що має кристалічні ґратки типу NaС1, забарвлений у коричневий колір. Оксид кадмію у воді не розчиняється, добре розчиняється у кислотах з утворенням солей, виявляє основні властивості.
Солі Кадмію здебільшого безбарвні; більшість із них у воді не розчиняється, за винятком Сd(NО3)2, СdС12, СdSО4, Сd I 2.
Під дією лугів на розчинні солі Кадмію утворюється гідроксид кадмію Сd(ОН)2, який виявляє основні властивості і, на відміну від Zn(ОН)2, в лугах практично не розчиняється. Сd(ОН)2 дещо розчиняється в дуже концентрованих розчинах лугів, добре розчиняється в кислотах. NН4ОН з йонами Сd2+ утворює осад Сd(ОН)2, здатний розчинятися в надлишку аміаку з утворенням досить стійкого безбарвного комплексного йона [Сd(NН3)4]2+ :
Сd(OН)2 + 4NH3 = [Сd(NH3)4]2+ + 2OH-
Здатність Кадмію до комплексоутворення вища, ніж Цинку. Для комплексних сполук Кадмію найхарактернішим є координаційне число 4 і тетраедричне розміщення лігандів. Проте Кадмій може також утворювати сполуки з координаційним числом 6. Йони Сd2+ здатні утворювати аквакомплекси, однак для Кадмію стійкішими є аміакати, галогеніди і псевдогалогеніди.
Завдяки здатності йонів Сd2 до утворення катіонних аквакомплексів типу [Сd(Н2О)4]2+ та [Сd(Н2О)6]2+ під час кристалізації розчинних солей Кадмію з водних розчинів утворюються кристалогідрати Сd(NO3)2 ∙ 4Н2О, Сd(СlO4)2 ∙ 6Н2О, 3СdSО4 ∙ 8Н2О.
Галогеніди кадмію можна добувати прямим синтезом, а також під дією на оксид кадмію галогеноводневих кислот. Галогеніди кадмію — слабкі електроліти. Хімічний зв'язок між атомами у молекулі галогеніду кадмію, як і взагалі в галогенідах елементів підгрупи Цинку, наближається до ковалентного. Безводні галогеніди СdГ2 добре приєднують аміак з утворенням сполуки CdГ2 ∙ 2NH3. Проте в разі надлишку аміаку найлегше утворюються комплексні сполуки з координаційним числом 4 або 6: [Сd(NНз)6]F2, [Сd(NН3)4]Вr2.
Під дією ціаніду калію на розчин солі Кадмію утворюється білий осад ціаніду кадмію Сd(СN)2, який здатний розчинятися у надлишку реактиву з утворенням досить міцного комплексного йона [Сd(СN)4]2- (Кнест= 1,4∙10 -19).
Сульфід кадмію СdS, що має жовте забарвлення, можна добути прямим синтезом (Кадмій має більшу спорідненість до Сульфуру, ніж до Оксигену) або під дією Н2S на розчини солей Кадмію. СdS не взаємодіє з розбавленими розчинами кислот. Сульфід кадмію розчиняється в гарячих хлоридній і сульфатній кислотах, найлегше розчиняється під час нагрівання в нітратній кислоті:
-2 +5 +2 0
3СdS + 8НNО3 = 3Cd(NO3)2 + 2NО↑ + 3S↓ + 4Н2О.
Сульфід кадмію вважається найкращою жовтою фарбою. Розчинні сполуки Кадмію отруйні.
Вміст Меркурію у земній корі становить 8,3 ∙ 10-6 %. Меркурій зрідка трапляється у вільному стані, а також у вигляді сульфідного мінералу червоного кольору — кіноварі НgS.
Відомо сім природних стабільних ізотопів Меркурію з масовими числами 196, 198—202, 204, найпоширенішими з яких є 200Нg (23,1 %) та 202Нg (29,8 %).
Ртуть добувають випалюванням кіноварі:
НgS+O2=Нg+S O2
Металічна ртуть утворюється відразу ж, оскільки НgO — сполука нестійка. Ртуть очищають від домішок промиванням 20%-м розчином НNO3; домішки (крім Аu і Аg) переходять у розчин. Ртуть високого ступеня чистоти добувають вакуумною дистиляцією або електролізом.
Світове виробництво ртуті становить близько 10 тис. т на рік.
Ртуть — єдиний метал, що за кімнатної температури перебуває у рідкому стані (температура плавлення -38,8 °С), вона має сріблястий блиск, летка. Пара ртуті дуже отруйна; при роботі з ртуттю слід бути дуже обережним!
Ртуть здатна розчиняти багато металів з утворенням рідких або твердих сплавів — амальгам. Часто під час взаємодії металів з ртуттю утворюються хімічні сполуки — інтерметаліди. Натрій з ртуттю утворює сім сполук різного складу.
Унікальні властивості ртуті роблять її надзвичайно важливою в ряді галузей техніки і в наукових дослідженнях. Багато ртуті витрачається для добування лугів і хлору (електроліз NaCl з ртутним катодом).
Амальгами натрію широко застосовують як відновник, амальгами срібла й олова — для пломбування зубів. Оскільки залізо з ртуттю амальгам не утворює, її можна зберігати в сталевому посуді.
За властивостями ртуть сильно відрізняється від цинку і кадмію. З металів підгрупи Цинку ртуть найменш активна. Внаслідок особливої стійкості 6s2-елек-тронної конфігурації зовнішнього електронного шару атомів Меркурію потенціал йонізації його атомів дуже високий. Тому на відміну від Цинку і Кадмію сполуки Меркурію здебільшого малостійкі. Меркурій — єдиний елемент, що утворює кластерний (багатоядерний) катіон Нgстійкий у водному розчині. Меркурій у сполуках, на відміну від Цинку і Кадмію, виявляє ступені окиснення +1 і +2. Сполуки зі ступенем окиснення +1 містять катіон Н g
На противагу сполукам Цинку, які не дуже отруйні, сполуки Меркурію надзвичайно отруйні!
Ртуть у ряду електрохімічних потенціалів розміщена після водню, отже. вона здатна розчинятися тільки в кислотах-окисниках. У цьому разі можуть утворюватись похідні як Нg(ІІ), так і Нg(I):
3Нg + 8НNO3 (розб.) = 3Нg(NO3)2 + 2NO↑ + 4Н2О;
6Нg+ 8НNO3 (розб.) = 3Нg2(NO3)2 + 2NO↑ + 4Н2О.
Сполуки Меркурію легко відновлюються. Під час відновлення Нg2+ спочатку утворюється Нg, а потім Нg0 :
2НgС12 + SnС12 = Нg2Сl2↓ + SnCl4;
Нg2С12 + SnС12 = 2Нg ↓ + SnС14.
Деякі сполуки Меркурію, що містять катіон Нg, нестійкі і в момент утворення відразу ж переходять у суміш сполук Меркурію(ІІ) і високодисперсної ртуті. Наприклад:
+1 +2 0
Нg(NO3)2 + 4КІ = К2[НgІ4] + НgІ + 2КNO3.
Гідроксид меркурію (ІІ) — сполука нестійка і розкладається на оксид меркурію(ІІ) і воду. НgO — основний оксид, однак його основні властивості виражені слабко. Тому більшість солей Меркурію(ІІ) здатні гідролізувати.
Порівняно з цинком і кадмієм, ртуть окиснюється гірше. Під час нагрівання ртуті до 300°С в атмосфері кисню утворюється оксид меркурію(ІІ) НgO. що має червоне забарвлення. За вищої температури НgО починає розкладатись на ртуть і кисень.
Однією з особливостей Меркурію є те, що для нього не відомі гідроксиди: під дією лугів на розчини солей Меркурію(І) утворюється осад Нg2O, а на розчин солей Меркурію(Н) — НgO.
Ртуть, як і її аналоги, виявляє підвищену активність до дії галогенів. У разі безпосередньої взаємодії ртуті з галогенами утворюються галогеніди Нg Г2 (Н2 Г2).
Хлорид меркурію(П), або сулема, НgСІ2 — це безбарвна речовина, яка порівняно мало розчиняється у холодній воді (6,6 г в 100 г Н2О за температури 20 °С) і добре у киплячій (58 г в 100 г Н2О). У молекулах галогенідів меркурію, як і в молекулах інших сполук, досить велика частка ковалентного зв'язку. Сулема НgС12 легко сублімується. Хлорид меркурію(ІІ) має молекулярні ґратки. На відміну від більшості солей інших металів, НgС12 є слабким електролітом, водні розчини цієї сполуки погано проводять електричний струм, гідролізують незначною мірою. Слабкими електролітами є також інші солі Меркурію(П), а Нg(СN)2 взагалі неелектроліт.
Хлорид меркурію(І), або каломель, Нg2С12, на відміну від НgС12, у воді не розчиняється. Його добувають у вигляді білого порошку під час нагрівання суміші НgС12 з ртуттю:
НgС12 + Нg= Нg2С12.
Під дією йодиду калію на солі Меркурію(І) утворюється оранжево-червоний осад йодиду меркурію(ІІ) НgI2, який розчиняється у надлишку йодиду калію з утворенням безбарвної комплексної солі К2[НgІ4]:
Нg12 + 2КІ = К2[НgІ4].
Аналогічні комплексні сполуки утворює Меркурій(ІІ) з йонами СN- і SCN-. Галогенідні і псевдогалогенідні комплексні сполуки Меркурію надзвичайно міцні. Найвищу здатність до комплексоутворення з галогенід- та псевдогало-генід-іонами виявляє Меркурій (порівняно з його аналогами).
Особливо активно ртуть взаємодіє з сіркою. Реакція відбувається навіть на холоді. Це можна пояснити великою міцністю зв'язку Нg — S і рідким агрегатним станом ртуті, що полегшує контакт з сіркою. Тому для зв'язування дрібних часточок розлитої ртуті на якійсь поверхні її обробляють сірчистим цвітом.
Сульфід меркурію НgS— досить стійка сполука. Вона не розчиняється в хлоридній кислоті, розчиняється тільки в царській воді під час кип'ятіння або в хлоридній кислоті за наявності окисника. Розчинення в царській воді Відбувається внаслідок утворення стійкого хлорокомплексу [НgСl4]2- :
-2 +5 +6 +2
3НgS+8HNO3+12HCl = 3H2[HgCl4]+3 H2SO4+8NO↑+4H2O
Роданід (тіоціанат) амонію з солями Меркурію(ІІ) утворює білий осад роданіду (тіоціанату) меркурію Нg(SCN)2. Осад розчиняється у надлишку роданіду амонію з утворенням комплексної сполуки (NН4)2[Нg(SCN)4].
Своєрідна реакція відбувається під час нагрівання роданіду (тіоціанату) меркурію(П): t
2Нg(SCN)2 = 2НgS + С3N4 + СS2.
Якщо запалити сіль Нg(SСN)2, починає утворюватися й горіти сірковуглець, а теплота, що виділяється при цьому, викликає розкладання решти солі. НgS, що має чорне забарвлення, і С3N4, забарвлений у жовтий колір, виділяються у вигляді дуже об'ємної пухкої маси незвичайної форми («фараонові змії»).
Порівнюючи катіони Нg2+ і Нg22+ за їх здатністю до комплексоутворення, легко помітити, що для Нg2+ вона значно вища, тому сполуки Меркурію(і) менш стійкі, ніж сполуки Меркурію(ІІ). Під дією окисників вони легко переходять у сполуки Меркурію(ІІ), а під час нагрівання або під дією надлишку активних лігандів піддаються диспропорціонуванню. Так, оксид меркурію(І) Нg2О частково розкладається вже за звичайної температури:
Нg2O = НgO + Нg,
а під дією надлишку йодиду калію на осад Нg2I2 відбувається реакція
+1 +2 0
Нg2І2+2КІ = К2[НgI4] + Нg.
Солі Меркурію(І) найкраще зберігаються у сухому вигляді. У розчині їх можна зберегти лише разом з металічною ртуттю.

 Ферум Fе, Кобальт Со та Нікол Ni знаходяться в бічній підгрупі VIII групи періодичної системи Д. І.Менделєєва. Їх властивості близькі одна до одної та суттєво відрізняються від властивостей інших шести елементів підгрупи. Тому їх об'єднують у сімейство Феруму, а дві інші тріади: Рутеній Ru — Родій Rh — Паладій Рl та Осмій Оs — Іридій Іr — Платина Рt — об'єднують у сімейство платинових металів.
Ферум Fе, Кобальт Со та Нікол Ni знаходяться в бічній підгрупі VIII групи періодичної системи Д. І.Менделєєва. Їх властивості близькі одна до одної та суттєво відрізняються від властивостей інших шести елементів підгрупи. Тому їх об'єднують у сімейство Феруму, а дві інші тріади: Рутеній Ru — Родій Rh — Паладій Рl та Осмій Оs — Іридій Іr — Платина Рt — об'єднують у сімейство платинових металів.
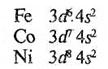 В основному стані атоми елементів тріади мають наступну будову зовнішніх шарів електронних оболонок:
В основному стані атоми елементів тріади мають наступну будову зовнішніх шарів електронних оболонок:
В утворенні хімічних зв'язків беруть участь 2 електрони s-підрівня (більш характерно) та d-електрони попередзовнішнього апідрівня (менш характерно).
Для атомів Fе та Со в сполуках характерні ступені окиснення +2 та +3, для Nі +2,рідко +3, +4. Це пояснюється тим, що з ростом заряду ядра притягання до нього електронів підсилюється в ряду Ре — Со — Ni. Це призводить до стабілізації ступеня окиснення +2. Для Феруму відомий ступінь окиснення +6 (К2Fе O4), який не відмічений у Со таNi.
 |
Fе, Со, Ni — сріблясто-білі метали з високими температурами плавлення, міцні та ковкі, усі вони феромагнетики.
За хімічною активністю елементи тріади феруму відносяться до металів середньої активності. При нагріванні й особливо в порошкоподібному стані вони досить енергійно взаємодіють з неметалами (О2, S, С12, Вr2, Р, Sі, В
і т. д.).
Розведені соляна та сірчана кислоти легко розчиняють Fе та Со, а Ni — при нагріванні (в ряду напруг металів Fе, Со, Nі стоять ліворуч Гідрогену). Концентровані Н2SО4 та HNО3 їх пасивують.
Для всіх елементів тріади характерне утворення комплексів з координаційним числом 6: Nа4[Fе(СN)6], [Со(NН3)6]С13, [Ni(NН3)б]С12.
До побічної підгрупи VIII групи належать елементи Ферум Fе, Кобальт Со, Нікель Ni, Рутеній Ru, Родій Rh, Паладій Pd, Осмій Оs, Іридій Іr, Платина Рt.
Атоми Fе, Ru і Оs мають (n-1)s2(п–1)р6(п-1)d6ns2-електронну конфігурацію зовнішніх електронних шарів, а атоми Ni, Fd і Рt — (п - 1)s2(п - 1)р6 (п - 1)d8ns2 або (n- 1)s2(п–1) р6(п- 1)d10ns0 Отже, за числом d-електронів атоми Ре, Ки, Оз подібні до атомів елементів підгрупи Мангану, а атоми Ni, Рd і Рt — до атомів елементів підгрупи Купруму. Цим і визначається хімічна природа зазначених елементів, тобто їхня хімічна активність повинна змінюватись у такій самій послідовності, як і в елементів побічних підгруп І та VII груп—послаблюватись зі збільшенням протонного числа атомів елементів.
У тріадах металів восьмої групи (Fе, Со, Nі; Ru, Rh, Рd; Оs, Іr, Рt) зліва направо внаслідок ефекту d -стиснення радіуси атомів дещо зменшуються, що зумовлює послаблення активності елементів.
Найактивнішими є Ферум, Рутеній і Осмій, які здатні утворювати сполуки з вищими ступенями окиснення, найпасивнішими серед елементів своїх рядів — Нікол, Паладій і Платина, що не утворюють сполук з високими ступенями окиснення. Активність елементів Осмію, Іридію і Платини крім ефекту d-стиснення визначається також ефектом лантаноїдного стиснення, тому ці елементи за властивостями дуже подібні до відповідних металів 5-го періоду.
Метали родини Феруму (залізо, кобальт, нікель) досить активні, на відміну від інших металів VIII групи, тому їх виділяють в окрему родину (фероїди), а метали двох інших тріад подібні між собою і до платини, тому їх об'єднують у родину платинових металів (платиноїди).
Відмінність у хімічній активності елементів родин Феруму і платинових металів позначилась також на їхній геохімічній характеристиці. В той час як метали родини Феруму перебувають лише у зв'язаному стані, платинові трапляються як в одних і тих самих рудах, так і"в самородному стані.
Деякі властивості елементів родини Феруму подано в табл. 5.2.3.
Таблиця 5.2.3.
Деякі властивості елементів та простих речовин родини Феруму
| Назва елемента | Хі-міч- ний сим-вол | Будова зов-нішнього і передостан нього елек- тронних шарів атома | Радіус атома, нм | Радіус йона E2+, нм | Енергія йоніза-ції Е → E+ еВ | Густина, г/см3 | Температура °С | Температура | Стандартний електродний потенціал процесу E2+→ E+, В | ||
| Ферум | Fе | 3d6 4s2 | 0,126 | 0,080 | 7,89 | 7.87 | -0,440 | ||||
| Кобальт | Со | 3d7 4s2 | 0.125 | 0,078 | 7,87 | 8,84 | -0,286 | ||||
| Нікол | Ni | 3d8 4s2 | 0.124 | 0,074 | 7,63 | 8,91 | -0.250 | ||||
Ферум — четвертий елемент (після О, Sі, Аl) за поширенням у земній корі (4,65 %). Іноді трапляється в природі у вільному стані (метеоритного походження).
Залізо добувають в основному з руд: магнітного залізняку Fе3О4, червоного залізняку Fе2О3, бурого залізняку Fе2О3 ∙ nH2О, сидериту FеСО3, піриту FеS2, арсенопіриту FеАsS.
Кобальт і Нікол менш поширені у природі: їх вміст у земній корі становить відповідно 3∙103 і 8∙10-3%. Прості речовини цих елементів (метали) трапляються в сплаві з залізом у метеоритах. Найважливішими мінералами Со і Ni є: кобальтин СоАsS, шпейсовий кобальт СоАs2, залізонікелевий колчедан (Fе, Ni9S8, нікелін NіАs, арсенонікелевий блиск NiAsS тощо.
Природний Ферум складається з чотирьох стабільних ізотопів. Кобальт у природі існує у вигляді одного стабільного нукліда 59Со, Нікол — у вигляді п'яти.
В організмі людини міститься близько 3г Феруму, здебільшого він входить до складу гемоглобіну крові. Недостатня кількість Феруму в організмі людини зумовлює недокрів'я. Кобальт теж є дуже важливим елементом, що забезпечує нормальну життєдіяльність живих організмів, він входить до складу кровотворного вітаміну В12.
Нікол, на відміну від Феруму і Кобальту, отруйний. Вважають, що Нікол у мізерних кількостях може відігравати роль каталізатора різних процесів у живому організмі.
Для технічних цілей потрібне в основному залізо, що містить близько 1 % вуглецю (сталь). Сталь виплавляють у два етапи: спочатку відновлюють залізну руду надлишком вуглецю і добувають сплав, що містить 3 – 4 % С (чавун), потім виплавляють сталь, видаляючи з чавуну надлишок вуглецю.
Чавун виплавляють у доменних печах, які мають форму зрізаного конуса і футеровані зсередини вогнетривкою цеглою. Висота печей досягає 30 м, внутрішній діаметр — близько 12м. На рис. 5.2.4 схематично зображено розріз доменної печі та зазначено основні стадії процесу виплавляння чавуну. Верхня половина печі (див. рис. 5.2.4,a)називається шахтою. Шахта закінчується отвором — колошником. Колошник закривається колошниковим затвором — рухомим конусом. Нижня частина доменної печі — горно, найширша, вона називається розпаром. Доменна піч працює за принципом протитечії: шихта рухається зверху вниз, а нагріті гази — знизу вгору.
У доменну піч шарами завантажують кокс і агломерат — відповідно підготовлену руду, спечену з флюсами. Через спеціальні отвори в горні (фурми) вдувають гаряче повітря або кисень для підтримування горіння і потрібної для виплавляння чавуну температури. У горні вугілля згоряє з утворенням СО2, який, проходячи крізь верхні шари розжареного коксу, перетворюється на СО. Оксид карбону(П) відновлює більшу частину руди, окиснюючись знову до СО2 (див. рис. 5.2.4,б)
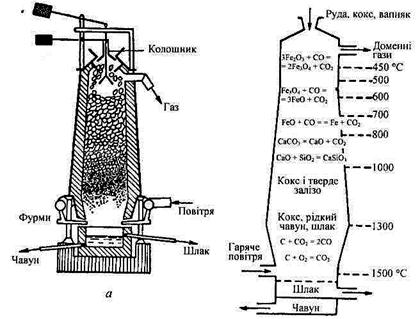
б
Рис. 5.2.4. Схема доменної печі (а)та хімічні реакції, що відбуваються в різних частинах доменної печі (б)
Окремі стадії процесу виплавляння чавуну можна виразити таким сумарним рівнянням: Fе2О3 + 3СО = 2Fе + 3СО2.
Процес відновлення залізної руди відбувається в основному у верхній частині шахти.
Оскільки пустою породою в руді є здебільшого тугоплавкий оксид силіцію (ІV) SіО2, то як флюс, що утворює з пустою породою порівняно легкоплавкий шлак, беруть СаСО3, який під час взаємодії з Si2 дає СаSіО3 у вигляді шлаку. Крім силікатів у шлак переходять сполуки Сульфуру та Фосфору, завдяки чому чавун звільняється від шкідливих домішок.
Під час відновлення руди залізо, що утворилося, поступово опускається в розпар, температура в якому значно вища, і розчиняє в собі вуглець, утворюючи чавун.
Так задута доменна піч безперервно працює протягом кількох років. У піч добавляють нові порції шихти в міру їх опускання. В сучасній доменній печі за рік можна виплавити близько 1 млн.т. чавуну.
Чавун дуже крихкий метал, що зумовлено великим вмістом у ньому вуглецю — 4—5 %.
Є кілька способів виплавляння сталі з чавуну. Невеликі кількості сталі виплавляють у конвертерах за конвертерним,або бесемерівським,способом, згідно з яким розплавлений чавун наливають у конвертер і продувають крізь метал повітря. Внаслідок перебігу процесу окиснення окиснюється частина С до СО2, деякі домішки (Р, S, Sі тощо) і частково залізо. Оксиди фосфору, реагуючи з СаО або МgO, що добавляються до шихти, утворюють шлак, який використовується як добриво.
За мартенівським способом виплавляння сталі чавун розплавляють у полу-меневій відбивній печі, в яку завантажують й, крім чавуну, стальний брухт, що потребує переплавки, і деяку кількість залізної руди. В піч вводять також попередньо нагріті повітря і паливо.
Під час згоряння палива температура в печі досягає 1800—1900 °С, що достатньо для розплавляння матеріалів, завантажених у піч. Щоб добути сталь певного складу, в розплав вводять різні добавки. Домішки вигоряють за рахунок кисню повітря.
Конвертерний спосіб значно дешевший і використовується ширше. Однак якість сталі, виплавленої бесемерівським способом, нижча, ніж мартенівської.
Значно прискорюється виробництво і підвищується якість металу у разі застосування кисню: на певних етапах конвертерного і мартенівського процесів крізь метал продувають чистий кисень або повітря, збагачене киснем.
Сучасним способом виплавляння сталі є виплавляння її в електричних печах.
Незалежно від способу виплавляння рідка сталь завжди містить деяку кількість розчиненого кисню (до 0,1 %), що призводить до погіршення механічних характеристик сталі. Тому процес виплавляння сталі завершують и розкисненням, добавляючи розкисники (манган, алюміній, силіцій, титан), які активно сполучаються з киснем.
Добре розкиснена сталь твердне без виділення газів, спокійно. Тверду сталь добувають у вигляді зливків.
На відміну від чавуну, сталь пластична, твердість її залежить від вмісту вуглецю: м'яка сталь містить до 0,3, а тверда — від 0,3 до 2,7 % С. М'яку сталь легко перетворити на ковке залізо.
Чисте залізо добувають у вигляді порошку відновленням його оксидів воднем або термічним розкладанням карбонілу феруму Fе(СО)5. Різні вироби з заліза виготовляють методом порошкової металургії. Чисте залізо містить до 0,0002 % домішок.
Процес добування кобальту і нікелю дещо складніший, ніж технічного заліза.
Під час випалювання арсеносульфідних руд утворюється суміш оксидів ніколу та кобальту з домішками оксидів інших металів. Продукти випалювання обробляють хлоридною кислотою і після фільтрування осаджують сульфіди важких металів (СuS, PbS, Ві2S3) сірководнем. У розчині залишаються хлориди ніколу, феруму, кобальту. Суміш розчинних солей фільтрують і обробляють хлорним вапном і вапняною водою; внаслідок чого осаджується кобальт у вигляді Со(ОН)3:
+2 +1 +3 -1
2СоС12 + Са(ОС1)С1 + Н2О + 2Са(ОН)2 = 2Со(ОН)3↓ + ЗСаС12.
Осад Со(ОН)3 відфільтровують, висушують, прожарюють, а оксиди Со2О3 і СоО, що утворились, відновлюють.
З фільтрату можна осадити Нікол. який окиснюється за вищих значень рН, ніж Кобальт. Для цього, крім хлорного вапна як окисника, до фільтрату додатково добавляють вапняну воду. Нікол також осаджується у вигляді Ni(ОН)3, який просушують, переводять в оксид і відновлюють.
Світове щорічне виробництво кобальту становить декілька десятків тисяч тонн, нікелю — сотні тисяч тонн.
Нові методи переробки кобальто-нікелевих руд грунтуються на обробці їх сумішшю Н2SО4 і НNO3 в автоклаві з наступним розділенням солей металів катіонним методом або екстракцією.
Залізо, кобальт, нікель мають сріблястий блиск, зберігають стійкість на повітрі до 400—700 °С завдяки наявності на їхній поверхні захисної оксидної плівки. Найстійкішим проти дії окисників є нікель, тому він широко використовується для антикорозійного покриття інших металів. Найменш стійким проти дії окисників є залізо. У високодисперсному стані Fе, Со, Ni пірофорні — здатні самозайматися на повітрі. Хімічно чисте залізо — м'який пластичний метал, м'якший за золото і срібло. Кобальт значно твердіший, ніж залізо і нікель.
У заліза, кобальту і нікелю різко виражена парамагнітність, вони притягуються магнітом, намагнічуються і тому називаються феромагнітними. Нікель має нижчу феромагнітність, ніж кобальт і залізо.
За густиною, температурами плавлення і кипіння метали родини Феруму дуже подібні між собою, вони пластичні і здатні проводити електричний струм.
Залізо існує у вигляді чотирьох алотропних видозмін (α-, β-, γ-, δ-залізо), кожна з яких має певний інтервал термодинамічної стійкості.
Наявність навіть невеликих кількостей домішок інших елементів у залізі, кобальті, нікелі призводить до різкої зміни механічних і фізико-хімічних властивостей цих металів. Крім цього, на властивості металів значною мірою впливає термічна і механічна обробка.
Залізо та його сплави становлять основу сучасної техніки. Нікол є однією з найважливіших легуючих добавок до сталей, залізо — основа чорної металургії. Кобальт почали застосовувати порівняно недавно у вигляді спеціальних сплавів: це сплави типу побідиту, що складаються з карбідів вольфраму і молібдену на кобальтовій основі. Постійні магніти виготовляють із сплаву заліза з алюмінієм, нікелем і кобальтом (сплав алніко), який має феромагнітні властивості. Широко використовуються жаростійкі сплави на основі нікелю (ніхром, що містить Nі, Сr тощо). Із мідно-нікелевих сплавів, наприклад, мельхіору, виготовляють монети, ювелірні прикраси.
Високомагнітні (містять 45—80 % Nі) та немагнітні (1—25 % N1) сталі, леговані хромом, використовують у приладобудуванні. Fе, Со, Ni та сполуки цих металів служать каталізаторами. Високодисперсний нікель каталізує процес гідрування органічних сполук, зокрема жирів.
Залізо, кобальт і нікол — метали середньої хімічної активності. Для них характерні ступені окиснення +2 і +3. Збільшення заряду ядра атомів цих елементів (посилення притягання до нього електронів) зумовлює стабілізацію ступеня окиснення +2 у разі переходу від Fе до Ni. Отже, з металів родини Феруму найлегше виявляє ступінь окиснення +3 Ферум. Нікол здебільшого виявляє ступінь окиснення +2.
Під час нагрівання метали родини Феруму здатні безпосередньо сполучатися з сіркою, галогенами, киснем, фосфором. Сухий хлор із залізом не взаємодіє, тому його зберігають у стальних балонах. За наявності вологи метали родини Феруму енергійно взаємодіють з хлором:
2Fе + 3Сl2 = 2FеС13; Со + Сl2 = СоСl2; Ni + Сl2 = NiС12.
З сіркою залізо, кобальт і нікель утворюють сульфіди МеS, що мають чорне забарвлення.
З воднем метали родини Феруму не утворюють стехіометричних сполук, однак вони здатні вбирати значні кількості водню, особливо в високодисперсному стані. Найкраще вбирає водень високодисперсний нікель, який утворює продукт, близький за складом до NіН2. Утворення таких металічних фаз зумовлює високу каталітичну активність металів родини Феруму (особливо нікелю).
За невисоких температур залізо, кобальт, нікель утворюють з азотом нітриди Fе2N, СоN, Nі3N2 тощо, проте в разі сильного нагрівання ці сполуки розкладаються.
Метали родини Феруму виявляють малу активність стосовно вуглецю. Ферум утворює Fе3С, який стійкий лише за високих температур, малостійкий карбід може утворювати Кобальт. Для Ніколу карбіди не добуті.
Для Fе, Со, Ni характерне утворення карбонілів. Карбоніли Fе(СО)5 і Со2(СО)8 добувають, діючи оксидом карбону(II) на порошкоподібні метали за підвищеного тиску і температури 100—200 °С. Нікель утворює сполуку Nі(СО)4 і за атмосферного тиску. Ферум, Кобальт і Нікель у карбонілах виявляють формальний нульовий ступінь окиснення.
Всі карбоніли дуже отруйні. Вони мають молекулярні кристалічні ґратки, практично не розчинні у воді, добре розчиняються в органічних розчинниках. Стандартні електродні потенціали φ°298 заліза, кобальту і нікелю відповідно дорівнюють -0,44, -0,28 і -0,25 В, отже, ці метали повинні легко розчинятися в кислотах-неокисниках з утворенням солей Ме2+ і Н2. Солі Ме3+ внаслідок перебігу цього процесу утворюватися не можуть, оскільки водень у момент виділення відновив би Ме3+ до Ме2+. Найлегше розчиняється у кислотах-неокисниках залізо, найважче — нікель. У концентрованих HNО3 і Н2SО4 залізо пасивується.
Метали родини Феруму стійкі проти дії розчинів і навіть розплавів лугів. У водному середовищі метали родини Феруму майже не кородують, най стійкі-ший проти корозії нікель. Стійкість заліза проти корозії залежить від ступеня його чистоти. Залізо високого ступеня чистоти не піддається корозії. Технічне залізо у вологому повітрі легко окиснюється і вкривається іржею.
Залізо, кобальт і нікель у подрібненому стані здатні самозайматися на повітрі за звичайних умов.
Найважливішими із сполук елементів родини Феруму є оксиди та солі. Добуто багато сполук, у яких елементи Fе, Со, Ni виявляють ступінь окиснення +2, ці сполуки стійкі у водних розчинах. Поряд з подібністю властивостей розглянутих елементів існує певна закономірність у їх зміні від Fе до Ni. У ряду Fе — Со — Ni внаслідок ефекту (d-стиснення зменшуються радіуси йонів, в результаті чого основні властивості гідроксидів у ряду Fе(ОН)2— Со(ОН)2—Nі(ОН)2 послаблюються, а стійкість комплексів зростає, що пов'язано також із заповненням електронами d -підрівнів з низькою енергією (октаедричне оточення лігандами).
Свіжодобутий Fе(ОН)2 відразу ж окиснюється киснем повітря:
4Fе(ОН)2 + О2 + 2Н2О – 4Fе(ОН)3.
Подібна реакція з Со(ОН)2 майже не відбувається, а для Ni(ОН)2 взагалі неможлива. Добути Ni(OH)3 можна тільки під дією дуже сильних окисників на Ni(OH)2.
Отже, відновна здатність сполук Ме2+ в ряду Fе – Со – Ni різко послаблюється.
Оксиди елементів (ІІ) родини Феруму добувають термічним розкладанням оксалатів відповідних металів. Сполуки FеО, СоО і NiO — основні оксиди, з водою не взаємодіють. Відповідні їм основи мало розчинні у воді. Добувають їх під дією лугів на розчинні солі Ме2+. Оксиди елементів (ІІ) родини Феруму і відповідні їм гідроксиди розчиняються в сильних кислотах з утворенням солей. Солі Fе2+ подібні до солей Мg2+, що зумовлено майже однаковими радіусами їхніх йонів.
У водних розчинах йони Fе2+, Со2+, Ni2+ утворюють октаедричні гідратні комплекси [E(Н2О)6]2+, які виділяються у вигляді кристалогідратів, наприклад: FеСl ∙ 6Н2О, СоС12 ∙ 6Н2О, NiCl2 ∙ 6Н2О, а також FеSО4 ∙ 7Н2О. Кристалогідрати Ніколу забарвлені в зелений колір, Кобальту — в рожевий, Феруму — в світло-зелений. Таке саме забарвлення мають розчини цих сполук.
Для Со2+, крім координаційного числа 6, характерне також координаційне число 4 з тетраедричним оточенням лігандами. Координаційні сполуки Со2+ з координаційним числом 4 мають яскраво-синє забарвлення.
Безводні солі Феруму та його аналогів за забарвленням відрізняються від кристалогідратів: СоС12 — синій, СоС12 ∙ 4Н2О — оранжевий, СоС12 ∙ 6Н2О — рожевий, СоSО4 — червоний, СоSО4 ∙ 7Н2О — рожевий, NiSO4 — жовтий, NiSO4 ∙ 7Н2О — яскраво-зелений тощо.
Солі Феруму (П), Кобальту (ІІ) і Ніколу (П), утворені слабкими кислотами та кислотами середньої сили (карбонати, ціаніди, силікати, фосфати, фториди тощо), мало розчиняються у воді, добре розчиняються у сильних кислотах. Карбонат феруму (ІІ) розчиняється в розчинах, що містять оксид карбону (ІV), з утворенням розчинних гідрогенкарбонатів:
FеСО3+ СO2 + Н2О = Fе(НСО3)2.
Крім молекул води йони Fе2+, Со2+, Ni2+ здатні координувати навколо себе молекули аміаку, амінів та інших сполук з утворенням катіонних або нейтральних комплексів. У ряду Fе—Со—Nі здатність до утворення комплексних аміакатів [Ме(NН3)6]2+ посилюється.
Нікол утворює найстійкішу, порівняно з Fе2+ і Со2+, комплексну сполуку [Ni(NН3)6]2+ блідо-фіолетового кольору. Завдяки підвищеній стійкості [Ni(NН3)6]2+ гідроксид ніколу (ІІ) добре розчиняється в гідроксиді амонію, що використовують у гідрометалургії для відокремлення Ніколу.
Йони Ме 2+ легко утворюють комплексні сполуки з псевдогалогенід-іона-ми. У разі надлишку SСN– -іонів Fе2+, Со2+, Ni2+ утворюють комплексні сполуки [Ме(SСN4]2– і навіть [Ме(SСN)6]4– . Найважливішою з них є комплексна сполука Кобальту, що має синє забарвлення. Цю реакцію використовують для аналітичного виявлення йонів Со2+.
Найстійкішими комплексними сполуками Fе2+, Со2+, Ni2+ є ціанідні. Саме тому внаслідок добавляння надлишку ціанід-іонів оксиди Ме(СN)2 швидко розчиняються: Fе(СN)2 + 4КСМ= К4[Fе(СN)6].
У лабораторній практиці широко використовується гексаціаноферат(ІІ) калію К4[Fе(СN)6] ∙3Н2О (жовта кров'яна сіль). Комплексний йон [Fе(СN)6]4– надзвичайно стійкий (Kнест [ Fе(СN) ]
] = 10 –35). Цій солі відповідає комплексна
= 10 –35). Цій солі відповідає комплексна
кислота Н4[Fе(СN)6] — біла_ тверда речовина, існує у вільному стані, належить до сильних кислот. Йон [Fе(СNб]4– має октаедричну будову. Гекса-ціаноферат (ІІ) калію здатний осаджувати йони Fе3+. При цьому утворюється сполука Fе4[Fе(СN)6]3 синього кольору, яка під назвою берлінська блакить використовується як фарба:
4FеС13 + ЗК4[Fе(СN)6] = Fе4[Fе(СN6]3↓+ 12КС1.
За цією реакцією аналітично виявляють йони Fе3+. Дуже подібні до координаційних сполук, що містять комплексні аніони, подвійні сульфати. Найпоширенішою з них є сіль Феруму й амонію (NН4)2SО4 ∙ FеSО4 ∙ 6Н2О (сіль Мора). Цю сіль часто використовують як джерело йонів Fе2+.
Як зазначалось, серед металів родини Феруму ступінь окиснення +3 найлегше виявляє Ферум.
Сполуки Fе3+ добувають під дією окисників на металічне залізо або окис-ненням сполук Феруму (ІІ). Під дією хлору на залізо утворюється хлорид феруму (Ш).
Особливо легко процес окиснення відбувається в лужному середовищі: Fе(ОН)2 в момент утворення починає переходити у Fе(ОН)3. Проте солі Феруму (П) здатні окислюватися і в кислому середовищі:
10FеSО4 + 2КМnО4 + 8Н2SО4 = 5Fе2(SО4)3 + К2SО4 + 2МnSО4 + 8Н2О.
Солі Феруму(ІІ) під час зберігання на повітрі окиснюються.
Солі Кобальту(ІІ) стійкі, окиснюються лише в лужному середовищі, а солі Ніколу — тільки в сильнолужному середовищі. Під час окиснення солей металів (ІІ) у лужному середовищі утворюються малорозчинні гідроксиди: бурий Fе(ОН)3, коричнево-бурий Со(ОН)3 і чорний Ni(ОН)3. Формули гідроксидів Ме(ОН)3 умовні, їх слід записувати так: Ме2О3 ∙ пН2О.
Гідроксид ніколу (ІІІ) Ni(ОН)3 є досить сильним окисником.
Під час нагрівання Fе(ОН)3 утворюється червоно-коричневий оксид феруму (ІІІ) Fе2О3, який досить стійкий і розкладається лише за температури понад 1400°С до Fе3О4. В разі зневоднення Со(ОН)3 утворюється Со3О4 (не Со2О3), а потім СоО. Гідроксид ніколу (ІП) розкладається за температури 140°С з утворенням NiO.
Обережно нагріваючи Со(NО3)2 та Ni(NО3)2, можна добути Со2О3 (темно-коричневий) і Ni2О3 (сіро-чорний);
+2 +5 -2 +3 +4 0
4Мe(NO3)2 = 2Ме2О3+ 8NO2↑ + О2↑.
Оксид і гідроксид кобальту(ІП) — сильні окисники:
+3 -1 +2 +2
2Со(ОН)3 + 6НС1 = 2СоСl2 + С12 ↑+ 6Н2О.
Гідроксиди Мe(ОН)3 погано розчиняються у воді. їхня основна функція виявляється значно слабкіше, ніж у Ме(ОН)2. Як оксиди, так і гідроксиди металів є амфотерними. Амфотерні властивості Fе (ОН)3 виявляються тільки під час сплавляння з лугами й основними оксидами. Процес супроводжується утворенням феритів:
Fе2О3 + 2NaОН = 2NaFеО2 + Н2О.
Під дією води ферити повністю гідролізують:
NaFеО2 + 2Н2О = Fе(ОН)3↓ + NаОН.
Амфотерні властивості Со(ОН)3 і Nі(ОН)3 виявлені значно слабкіше, ніж у Fе(ОН)3.
Оскільки сполуки Fе, Со і Ni, де ці елементи виявляють ступінь окиснення +3, є окисниками, то добути їхні сульфіди Ме2S3 шляхом осадження з водних розчинів неможливо. Під дією Н2S на розчин солей металів (ІП) утворюється суміш МеSі S.
Окиснювальна здатність йонів Ме3+помітно зростає від Феруму до Ніколу, тому й гідроксиди Ме(ОН)3по-різному взаємодіють з кислотами. Як уже зазначалось, Со(ОН)3, а тим більше Ni(OH)3, виступають окисниками відносно кислот і утворюють солі двовалентних металів і кисень.
Fе(ОН)3, взаємодіючи з кислотами, здатний утворювати стійкі солі сильних, багатьох середніх і навіть слабких кислот. Солі сильних кислот здебільшого добре розчиняються у воді і кристалізуються з молекулами води:
Fе(NO3)3 ∙ 9Н2О; Fе(SО4)3 ∙ 18Н2О; FеС13 ∙ 6Н2О тощо.
Солі Феруму(ІП) як солі дуже слабкої основи сильно гідролізують, внаслідок чого їхні розчини набувають бурого забарвлення:
[Fе(Н2О)6]3+ + Н2О 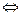 [Fе(ОН)(Н2О)5]2+ + Н3О+.
[Fе(ОН)(Н2О)5]2+ + Н3О+.
Якісною реакцією виявлення йонів Fе3+ є утворення забарвлених у червоний колір роданідних комплексів від [Fе(SСN)(Н2О)5]2+ до [Fе(SCN)6]3–.
Ферум(Ш) і Кобальт(Ш) утворюють дуже стійкі ціанідні комплекси [Fе(СN)6]3– і [Со(СN6]3–. Серед них найпоширенішим є гексашаноферат(ІІІ) калію К3[Fе(СN)6] (червона кров'яна сіль). Комплексний йон [Fе(СN)6]3– надзвичайно стійкий (Kнест [ Fе(СN)]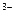 = 10 –42).
= 10 –42).
Під дією розчину К3[Fе(СN6] на розчини, що містять йони Fе2+, утворюється так звана турнбулева синька:
3FеСl2 + 2К3[Fе(СN)6] = Fе3[Fе(СN)6]2 + 6КСl.
В той час як бінарні сполуки для Со(ІП) нехарактерні, ступінь окиснення +3 для Со у комплексних сполуках стійкий. Комплексні сполуки Кобальту(Ш) добувають окисненням сполук Кобальту(ІІ):
СоС12 + 7КNO2 + 2СН3СООН =
= К3[Со(NО2)6]↓+ NO↑ + 2КС1 + Н2О + 2СН3СООК.
На відміну від простих солей, комплексні солі Феруму(ІІІ) та Кобальту(ІІІ) практично не гідролізують і значно важче відновлюються. Тривалентні метали здатні утворювати також фторидні комплексні сполуки К3[NiF6], К3[СоF6], Na3[FеF6], а Ферум також і хлоридні Me[FеСl4] та Ме2[FеСl5]. Особливо стійкими є фторидні комплексні сполуки Феруму(ІІІ), тому гідроліз сполук Ре3+ за наявності йонів F– послаблюється.
Крім ступенів окиснення +2 і +3 Ферум може виявляти також ступінь окиснення +6. Під дією сильних окисників у лужному середовищі на Ре(ОН)з або Fе2О3 утворюються ферати, наприклад:
+3 0 +6 -1
2Fе(ОН)3 + 3Вr2 + 10КОН = 2К2FеО4 + 6КВr + 8Н2О.
Сполуки Со (VІ) і Ni (VІ) не добуті.
Ферати лужних металів здатні розчинятися у воді з утворенням розчинів, забарвлених у червоний колір. Погано розчиняються у воді ферати лужноземельних металів. Ферати дуже сильні окисники, сильніші, ніж КМnО4, тому вони погано зберігаються, особливо їх розчини.
Сполуки Кобальту і Ніколу отруйні.
До побічної підгрупи IV групи належать елементи Титан Ті, Цирконій Zr, Гафній Hf і Резерфордій Rf. Всі вони повні електронні аналоги. Атоми елементів підгрупи Титану мають незавершений передостанній електронний шар, два d-електрони та два s-електрони в зовнішньому електронному шарі.
Титан, Цирконій і Гафній в разі утворення стійкої електронної структури втрачають свої електрони, тому для них основними є позитивні ступені окис-нення. Деякі властивості елементів підгрупи Титану наведено в табл. 5.2.5. Від Титану до Цирконію атомні та йонні радіуси елементів дещо збільшуються, а радіуси атомів і йонів Цирконію і Гафнію внаслідок лантаноїдного стиснення майже однакові. Тому за властивостями Цирконій і Гафній дуже подібні.
На відміну від елементів підгрупи Германію, в підгрупі Титану із зростанням протонного числа атомів елементів стійкість вищих ступенів окиснен-ня посилюється. Для Титану та його аналогів найхарактерніший ступінь окиснення +4, однак відомі також сполуки Титану(Ш) і Титану(ІІ). Цирконій і Гафній майже не виявляють здатності до утворення сполук з нижчими ступенями окиснення елементів.
Таблиця 5.2.5.
Читайте також:
- Алгоритми групи KWE
- Важливою ознакою класифікації є принцип побудови перетворювачів кодів, згідно з яким їх можна поділити на чотири групи.
- Варіанти групи крові
- Вимоги до керівника групи
- Вимоги до професійних і особистісних якостей керівника групи АСПН
- Витрати, пов'язані з одержанням освіти, можна розділити на три групи
- Відмінювання іменників другої відміни. Особливості поділу на групи іменників з основою на –р. Особливості відмінкових закінчень іменників другої відміни родового відмінка.
- Відновлення металів може проводитись різними методами, які зручно об'єднати в такі групи: пірометалургійні, гідрометалургійні та електрометалургійні.
- Віковий підхід і взаємодія різних рівнів соціального досвіду в діяльності різновікових дитячих об’єднань. Функції різновікової групи.
- Властивості елементів підгрупи берилію
- Вплив групи однолітків на формування особистості дошкільника.
- Вплив чисельності групи на її організованість і психологічний клімат
| <== попередня сторінка | | | наступна сторінка ==> |
| Побічної підгрупи І групи | | | Деякі властивості елементів та простих речовин побічної підгрупи IV групи |
|
Не знайшли потрібну інформацію? Скористайтесь пошуком google: |
© studopedia.com.ua При використанні або копіюванні матеріалів пряме посилання на сайт обов'язкове. |